



June 11, 2020 -- What makes the SARS-CoV-2 virus so virulent? Researchers from the U.S. National Institutes of Health (NIH) analyzed the genomics of the virus -- and compared it to other coronaviruses -- in a June 10 article in the Proceedings of the National Academy of Sciences.
The researchers used comparative genomics and machine learning to differentiate aspects of pathogenic SARS-CoV-2, SARS-CoV, and MERS-CoV from less pathogenic coronaviruses. SARS-CoV-2 is the seventh member of the Coronaviridae family known to infect humans. Together with SARS-CoV and MERS-CoV, these coronaviruses cause disease outbreaks associated with high mortality rates.
These highly pathogenic coronaviruses originated from zoonotic transmissions from animal hosts to humans. By contrast, other coronaviruses such as human coronavirus (HCoV)-HKU1, HCoV-NL63, HCoV-OC43, and HCoV-229E are endemic and cause seasonal common colds.
To determine the shared genomic determinants that cause high pathogenicity of coronaviruses, the researchers from the National Library of Medicine (NLM), part of the NIH, combined genome comparison techniques with advanced machine-learning methods. This approach was applied to specific key replication protein domains of all known coronaviruses with complete genomes.
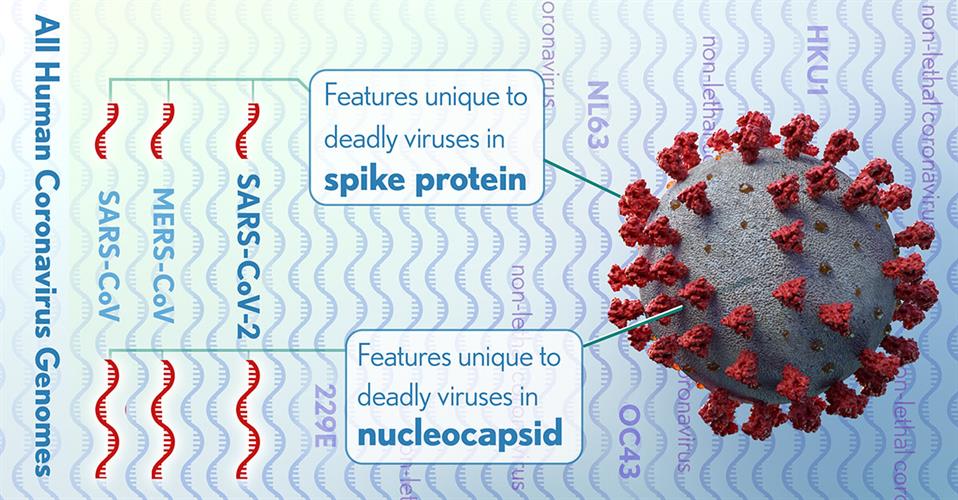
The full genomes of all human coronaviruses were aligned to identify regions (red) that might code for lethal differences in the virus that causes COVID-19, as well as severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS). These differences could be targets for testing or treatments. Image courtesy of Donny Bliss, National Library of Medicine.
The NIH researchers detected 11 regions of nucleotide alignments that were reliable in predicting highly pathogenic coronaviruses. These regions were contained in four proteins within the nucleocapsid and the spike glycoprotein, two of which were significantly enriched with these specific conserved sequences.
Specifically, the researchers determined that deletions, insertions, and substitutions of the nucleocapsid protein are the result of nuclear localization signals (amino acid sequences, typically positively charged lysines or arginines, that tag a protein for import). The team suggested that the growing positive charge accumulation of the nucleocapsid protein could contribute to the increased pathogenicity of virulent coronaviruses.
The researchers also identified a four-amino acid insertion in the connecting region of the spike glycoprotein of SARS-CoV-2, SARS-CoV, and MERS-CoV, but not in any of the less pathogenic coronaviruses. The insertion increases the length and flexibility of the connecting region and may affect the fusion process and contribute to the pathogenicity of these coronaviruses.
Lastly, the NIH team explored genomic features that may be associated with how coronaviruses cross the species barrier to humans. To do this, they aligned the genomes of highly pathogenic coronaviruses to their closest nonhuman infecting relatives and searched for insertions or deletions that occurred before the zoonotic jump to humans.
They found independent insertions located in the virus's spike glycoproteins, specifically within the receptor-binding domain (RBD) within the receptor-binding motif (RBM) of SARS-CoV and SARS-CoV-2, and DPP4 in the case of MERS-CoV. The insertions, although unique, correspond to loops connecting the β-sheets and β-strands of the secondary structure and contain a proline-cysteine amino acid doublet.
The different locations result in distinct RBM conformations and different receptor specificity in human cells. The flexibility allowed by these insertions could allow the spike glycoprotein to be more malleable in binding to a receptor and therefore allow zoonotic transmission.
"In this work, we set out to identify genomic features unique to those coronaviruses that cause severe disease in humans," said lead author Eugene Koonin, PhD, NIH Distinguished Investigator in the intramural research program of NLM's National Center for Biotechnology Information, in a statement. "We were able to identify several features that are not found in less virulent coronaviruses and that could be relevant for pathogenicity in humans. The actual demonstration of the relevance of these findings will come from direct experiments that are currently getting underway."
Do you have a unique perspective on your research related to genomics or virology? Contact the editor today to learn more.
---

Copyright © 2020 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


