



June 24, 2021 -- Researchers have designed "ideal" functional proteins using a trial-and-error iterative process that combines computer design and lab experiments based on the principles of protein folding. This work, published in Nature Communications on June 24, paves the way for de novo design of large proteins with relevant biochemical functions.
The design of ideal protein structures is guided by a set of principles characterized by consistent local and nonlocal interactions and structures without internal energetic frustration. These principles include a set of design rules relating local backbone structures to supersecondary structure packing of α-helices on paired β-strands, which generate funnel-shaped landscapes by disfavoring non-native states. These rules are what have made de novo design of ideal protein structures possible.
The coming together of distant parts of the protein has the advantage of allowing a much broader range of geometries than possible in a local chain segment, allowing for the formation of functional sites. Many enzymes are composed of a core of a central β-sheet with five or more strands surrounded on both sides by α-helices.
The team of researchers from Japan and the U.S. who authored the paper had previously designed proteins with four-stranded αβ-proteins with different topologies, sizes and shapes, and larger TIM-barrels. TIM-barrels, also known as α/β barrels, consist of eight α-helices and eight parallel β-strands that alternate along the peptide backbone. However, these designs were too small to harbor active sites.
"The ideal proteins we have created so far are much more stable and more soluble than proteins commonly found in nature," explained co-author Rie Koga of Japan's National Institutes of Natural Sciences (NINS), in a statement. "We think these proteins will become useful starting points for designing new biochemical functions of interest."
The team also wanted to test the generality of their previous designs.
"We set out to test the generality of the design principles we developed previously by applying them to the design of larger α/β proteins with five and six β-strands," said co-author Nobuyasu Koga, PhD, associate professor in the Institute for Molecular Science (IMS) at NINS.
The results from the design of the larger proteins were puzzling to the researchers. The first three protein designs attempted by the team suggested something was missing in the energy function or design concept. They found that the experimental structures differed from computer models, resulting in proteins that folded differently by swapping the internal locations of β-strands. The swapped state had free energy that was favorable to the designed state, according to the researchers.
To explain this phenomenon, the team conducted iterative computational designs and experiments, where Rosetta sequence-independent folding simulations were used to generate backbone structures and subsequently build side chains on each of the generated protein structures. The designed proteins were then expressed, purified, and characterized by circular dichroism (CD) spectroscopy, size-exclusion chromatography combined with multiangle light scattering (SEC-MALS), and H-N heteronuclear single quantum coherence (HSQC) NMR spectroscopy.
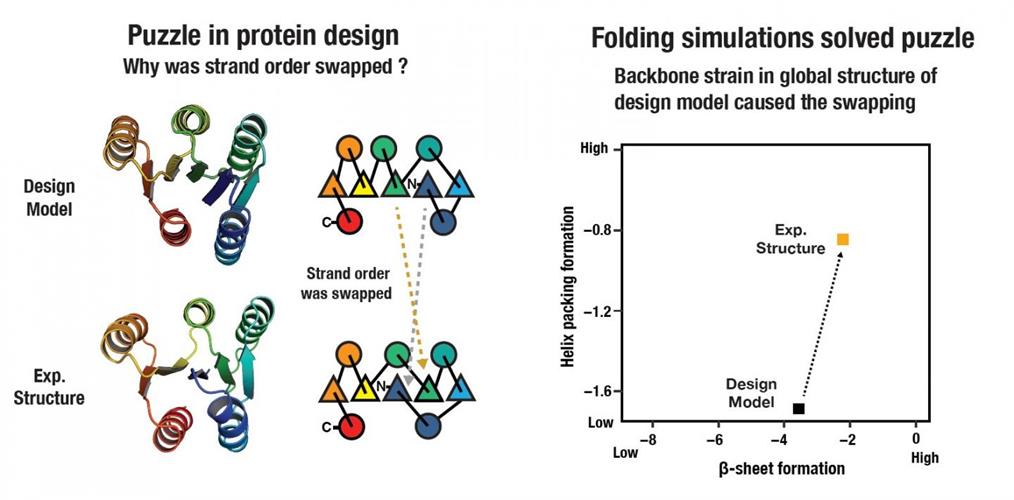
Left: the strand order swapping in de novo design of larger alpha-beta proteins has been a long-standing problem for the research team. Right: backbone ensembles generated from folding simulations identified that backbone strain caused the strand swapping. Image courtesy of NINS/IMS.
"We emphasize that experimental structure determination is important for iterative improvement of computational protein design," said co-author Gaohua Liu, PhD, chief scientific officer of Nexomics Biosciences.
Through this trial-and-error process, the scientists established that long-range backbone strain likely accounts for the favoring of the swapped state. They offered that the protein order requires consideration for not only local backbone strain associated with supersecondary structure formation but also backbone strain arising from incompatibility in global tertiary structure.
By modulating the design blueprints of the proteins to relieve frustration in β-sheet formation via shortening strand lengths, the authors were able to design proteins that fold into the original computer-generated formations.
"Sometimes we learn the most from these ideal proteins when their experimental structures differ, rather than match, their intended design, since this can lead to a deeper understanding of the underlying principles," added co-author Gaetano Montelione, PhD, professor of chemistry and chemical biology at Rensselaer Polytechnic Institute.
The results of the study illustrate the value of experimental structure determination in guiding de novo design of functional proteins. The work also stresses the importance of consistency between local, supersecondary, and global tertiary interactions in determining protein topology.
Next, the researchers plan to continue studying the trade-off between more functional proteins and with what could be considered less-than-ideal qualities.
"We would like to design proteins with more complex functional sites by incorporating non-ideal features such as longer loops, which are important not only for function but also for relieving global backbone strain," said co-author David Baker, PhD, professor of biochemistry at the University of Washington.
Do you have a unique perspective on your research related to protein design or molecular biology? Contact the editor today to learn more.
---
Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


