



October 14, 2020 -- A new approach to RNA sequencing can enable scientists to extract 10 times more information from a single cell, including gene expression and subtle differences between healthy and diseased cells. The study, published in Immunity on October 13, reveals the power of the improved Seq-Well method and provides evidence of its efficacy in five inflammatory skin diseases.
Single-cell RNA sequencing (scRNA-seq) is a powerful tool to characterize cells. Current scRNA-seq platforms, despite offering high throughput, are inefficient and provide low resolution among distinct cell states and molecular features. Improving the resolution of scRNA-seq can help define heterogenous populations of immune cells and subtle differences in surface receptors, transcription factors, and cytokine expression.
Most high-throughput scRNA-seq methods rely on barcoding of cellular components to recover single-cell transcriptomes for thousands of cells at once. This is achieved by isolating uniquely barcoded poly-dT oligonucleotides that can capture and tag cellular messenger RNA (mRNA) during reverse transcription.
In a second step, an additional oligonucleotide priming site is added to newly synthesized complementary DNA (cDNA) to enable polymerase chain reaction (PCR)-based amplification. The second step can be highly inefficient, leading to loss of molecules that have been converted to cDNA but not successfully tagged with a secondary PCR priming site.
A new method to reclaim resolution
Researchers from the Massachusetts Institute of Technology (MIT) developed Seq-Well S3 ("Second-Strand Synthesis") as a massively parallel scRNA-seq protocol that uses a randomly primed second-strand synthesis to recover cDNA molecules to facilitate template-switching. This generates double-stranded cDNA that is labeled on one end with the SMART sequence and its reverse complement on the other, making it more accessible for PCR enzymes to amplify the molecules.
From earlier versions of the method, the updated Seq-Well S3 obtained a 5- to 10-fold increase in the number of unique genes and transcripts captured per cell. The method also facilitated enhanced detection of lineage-defining factors in immune and organ-specific parenchymal cells -- such as transcription factors, cytokines, and cytokine receptors -- that are often transiently or lowly expressed.
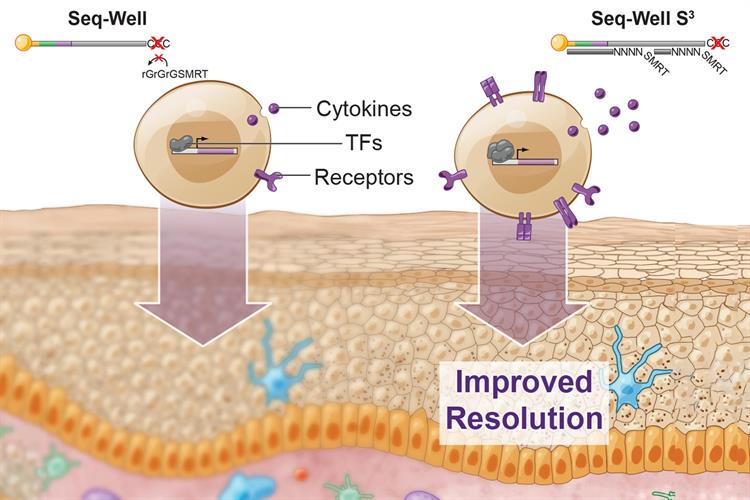
MIT researchers have greatly boosted the amount of information that can be obtained using Seq-Well, a technique for rapidly sequencing RNA from single cells. Image courtesy of MIT.
"We were able to vastly improve the amount of per cell information content with a really simple molecular biology trick, which was easy to incorporate into the existing workflow," explained lead author Travis Hughes, MD/PhD student in the Harvard-MIT Program in Health Sciences and Technology, in a statement. The protocol can be integrated into existing bead-based RNA-seq platforms (Drop-Seq, Slide-Seq), and the researchers have made it available to the broader research community.
Defining heterogeneity of diseased cells
It has been established that many diseased and healthy cells have heterogeneous immune and parenchymal cell populations. So, the researchers used Seq-Well S3 to examine the cellular composition of normal skin and uncover alterations in cellular abundance and phenotype across multiple inflammatory skin conditions, including acne, alopecia areata, granuloma annulare, leprosy, and psoriasis.
"It's become clear that these technologies have transformative potential for understanding complex biological systems. If we look across a range of different datasets, we can really understand the landscape of health and disease, and that can give us information as to what therapeutic strategies we might employ," says Alex Shalek, PhD, associate professor of chemistry at MIT.
In total, the researchers processed 19 skin biopsies and retained over 38,000 high-quality single-cell transcriptomes using Seq-Well S3. They were able to recover 15 primary cell types. To further define biological features, the team used the method to examine subpopulations of T cells, myeloid cells, endothelial cells, dermal fibroblasts, and keratinocytes in each inflammatory condition. The team found, for example, regulatory T cells, dysfunctional NR4A1-expressing T cells, and senescent SESN3+ T cells were over-represented, potentially reflecting T-cell dysfunction in psoriasis pathology.
The team also distinguished patterns associated with multiple diseases by looking across different inflammatory skin conditions, revealing common and unique features. For instance, they found that a group of natural killer cells, γΔ T cells, and a sub-cluster of immature cytotoxic T cells are derived from leprosy and granuloma annulare, indicating common T-cell programming in both forms of inflammation.
The results from the study created a draft atlas of human skin inflammation and a compendium of cell types and states that can be used by the broader research community.
"This is by no means an exhaustive compendium, but it's a first step toward understanding the spectrum of inflammatory phenotypes, not just within immune cells, but also within other skin cell types," Hughes noted.
"You never know what you're going to want to use these datasets for, but there's a tremendous opportunity in having measured everything," Shalek added. "In the future, when we need to repurpose them and think about particular surface receptors, ligands, proteases, or other genes, we will have all that information at our fingertips."
The technique is being used to study other diseases and cell types such as tuberculosis, malaria, HIV, Ebola, and food allergies.
Do you have a unique perspective on your research related to proteomics or genomics? Contact the editor today to learn more.
---
Copyright © 2020 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


