



October 25, 2022 -- Researchers at New York University (NYU), in collaboration with scientists at the biopharmaceutical company AbbVie, have developed a new computational tool that can quickly and efficiently map the position of water molecules within crystal structures. Their findings could be used during the drug development and discovery process to predict which crystals are likely to include water and anticipate the different possible structures of a given drug formulation.
Molecular crystals can take on water either during the crystallization process or by absorbing it from the environment. When this occurs, the water can alter the crystals' properties, such as improving or impairing their ability to dissolve.
These water-holding crystals, known as crystal hydrates, represent a significant portion of patented drugs, and about one-third of commercially available drugs have active ingredients in hydrated form. Predicting which crystals are likely to contain water and at what level has been difficult and computationally intensive.
In their study, published October 17 in the journal Proceedings of the National Academy of Sciences, the researchers developed a computational protocol -- called Mapping Approach for Crystal Hydrates (MACH) -- that can quickly determine whether a given compound will likely form a crystal hydrate.
They also created a system for predicting both stoichiometric and non-stoichiometric hydrates. The former have a defined ratio of water molecules to other molecules and are easy to predict while the latter do not and are hard to predict.
MACH establishes a set of rules to systematically determine where water would likely be inserted into a crystal based on the unique structure of and chemical environment within each crystal. Its algorithm first instructs the computer to build a crystal structure in its dry form. It then tests to see if water will fit into the dry framework by overlaying a liquid water sample onto the crystal structure.
Next, MACH considers additional rules for how water will interact with the host crystal, further reducing the number and location of water molecules that can be incorporated into the crystal. These steps are then repeated many times with different configurations of water molecules.
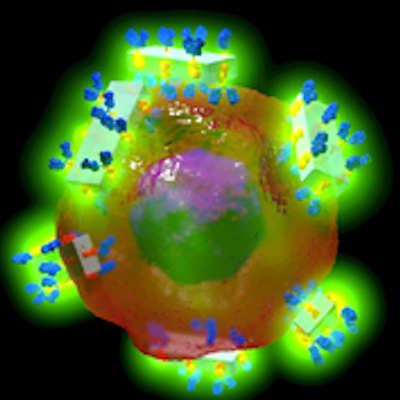
At the end, the researchers are left with a map of the remaining water molecules, illustrating which crystals are likely to form hydrates -- both stoichiometric and non-stoichiometric -- and where the water molecules are incorporated.
The researchers demonstrated the ability of MACH to provide an accurate mapping of water molecules in three drugs: a plant-derived compound named brucine, the antidepressant paroxetine hydrochloride, and the diabetes drug sitagliptin tartrate. The crystal structures of each drug have structural voids of different sizes and chemical environments.
"Given its simplicity and speed, MACH offers a promising tool for efficiently predicting hydrates and could be integrated into drug development and formulation workflows to build a more complete landscape of possible crystal structures," Mark Tuckerman, PhD, chair of the department of chemistry at NYU and the study's senior author, said in a statement.

Copyright © 2022 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


