



July 1, 2021 -- A new software tool can help researchers further utilize single-cell RNA sequencing (scRNA-seq) data to learn more about individual cells by incorporating biologically relevant data into the analysis. The machine-learning algorithm, detailed in Genome Research on June 30, can be used to predict which transcription factors are most active in individual cells.
There are hundreds of transcription factors inside human cells, and it can take years of trial-and-error experimental research to identify which are turned on or off in specific cell types. This is important information as these proteins could be leveraged as potential drug targets.
Previous methods developed to infer transcription factor activities have been based on messenger RNA (mRNA) encoding the transcription factors. These strategies may not be representative of true biological functions because the activity of transcription factors is often regulated at the post-translational level and key changes to transcription factors may not be detected at the mRNA level.
ScRNA-seq is a powerful tool for investigating the transcriptomic heterogeneity among cells. However, elucidating the underlying biological functions and regulatory mechanisms (i.e., transcription factor activities or gene regulatory networks) of cells based on these data is challenging due to difficulties in the integration of biological context data.
Most applications of scRNA-seq have been focused on spatial biology, which aims at identifying cell clusters based on proximity of individual cells in low dimensional space. However, this analysis does not take into consideration biological function and does not help researchers uncover regulatory mechanisms in subpopulations of cells.
"One of the challenges in the field is that the same genes may be turned 'on' in one group of cells but turned 'off' in a different group of cells within the same organ," said Dr. Jalees Rehman, professor at the University of Illinois Chicago, College of Medicine. "Being able to understand the activity of transcription factors in individual cells would allow researchers to study activity profiles in all the major cell types of major organs such as the heart, brain or lungs."
Improving scRNA-seq analysis
To address the limitations of previous scRNA-seq analysis strategies, a team of University of Illinois Chicago researchers led by Rehman and Yang Dai, PhD, an associate professor of bioinformatics at the university, developed the Bayesian inference transcription factor activity model (BITFAM).
The system combines new gene expression profile data gathered from scRNA-seq with existing biological data on transcription factor target genes. The researchers associated transcription factors with their predicted target gene set, obtained from GTRD databases of chromatin immunoprecipitation sequencing (ChIP-seq) that contains over 17,000 transcription factor samples.
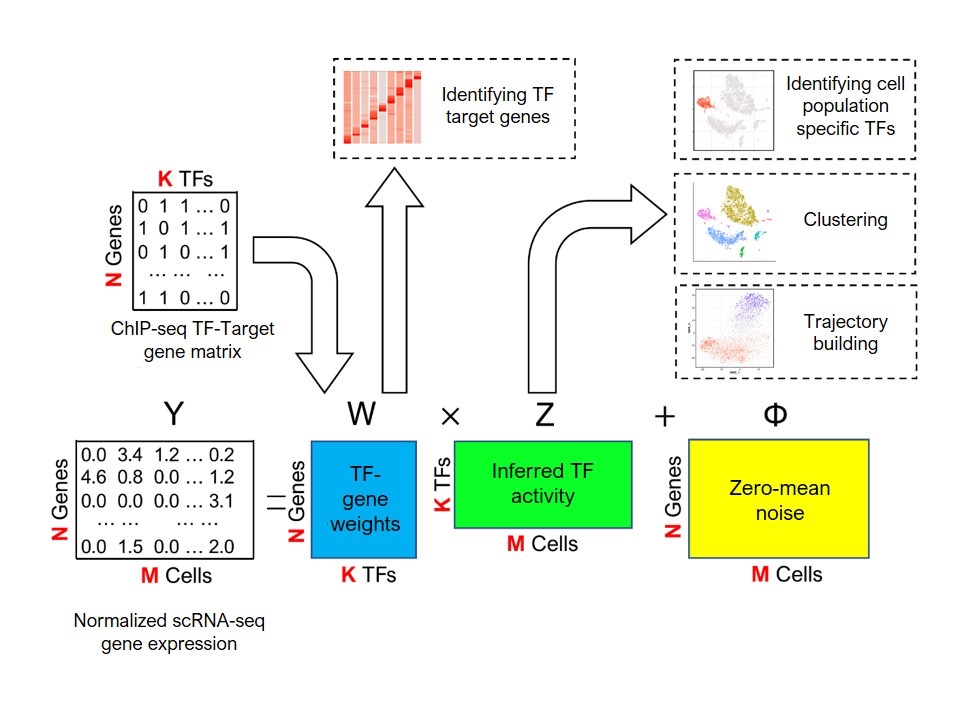
A schematic overview of the BITFAM machine-learning system developed by researchers at UIC. User-provided sequencing data ("Normalized scRNA-Seq gene expression") and existing data on transcription factor binding sites ("ChIP-seq TF-Target gene matrix") are analyzed to predict transcription factor activity ("Inferred TF activity") that can be leveraged for a broad range of analyses. Image courtesy of Genome Research. Licensed under CC BY 4.0.
The model integrates prior biological knowledge (ChIP-seq) with observed data to infer transcription factor activities in cell subpopulations. With this information, the system runs numerous computer-based simulations to find the optimal fit and predict the activity of each transcription factor in the cell.
They applied the model to several scRNA-seq datasets including a mouse dataset that contains information on all major organs during adult homeostasis, a blood cell development dataset, and an interference CRISPR dataset with 50 targeted CRISPR transcription factor deletions.
In these contexts, BITFAM was able to infer biologically meaningful transcription factor activities of selected well-established transcription factors with known biological functions. For instance, it predicted the high activity of T-cell acute lymphocytic leukemia protein 1 (TAL1) in lung endothelial cells, which matches previous knowledge that TAL1 is an important factor for endothelial gene activation.
"Our approach not only identifies meaningful transcription factor activities but also provides valuable insights into underlying transcription factor regulatory mechanisms," said first author Shang Gao, a doctoral student in the department of bioengineering at the University of Illinois Chicago. "For example, if 80% of a specific transcription factor's targets are turned on inside the cell, that tells us that its activity is high. By providing data like this for every transcription factor in the cell, the model can give researchers a good idea of which ones to look at first when exploring new drug targets to work on that type of cell."
The authors also noted that BITFAM could be used to discover novel heterogenous subpopulations with subtle phenotype differences driven by regulation of transcription factors.
"This new approach could be used to develop key biological hypotheses regarding the regulatory transcription factors in cells related to a broad range of scientific hypotheses and topics," Dai said. "It will allow us to derive insights into the biological functions of cells from many tissues."
The researchers said that the new system is publicly available and could be applied widely because the model can easily be combined with additional analysis methods that may be best suited for their studies, such as finding new drug targets.
Do you have a unique perspective on your research related to bioinformatics or cell biology? Contact the editor today to learn more.
---


Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


