



July 14, 2018 -- The benefits of polyclonal antibodies as tools for assay-specific target discovery and detection are numerous. As the future of basic research, diagnostics and biomarker discovery is dependent on high-quality reproducible data, there is a need to understand the importance of and benefits of these valuable tools.

All antibody forms — polyclonal, hybridoma-based monoclonal and recombinant monoclonal — have pros and cons for development, validation and use. Yet, polyclonal antibodies are embroiled in a firestorm of controversy concerning data reproducibility. We address best practices for developing and using polyclonal antibodies, pitfalls to their use and how to avoid them, and benefits to the life science community. Eliminating their use risks overlooking the unique benefits of polyclonal antibodies as 'fit-for-purpose' life science tools.
Polyclonal antibodies: caught up in the conflagrationAntibodies are critical reagents most often used by life science and translational medicine researchers. Thoughtful antibody design and development allows for these tools to be used in many different applications, techniques and instrumentation. All forms of antibodies, polyclonal (pAb), hybridoma-based monoclonal (mAb) and recombinant monoclonal (rAb), have both pros and cons as research tools and have attributes that differentiate them from one another. pAbs display multi-epitope binding properties, which make these reagents ideally suited for many applications, whereas their finite supply detracts from their utility. As inherently limited supplies of pAbs become exhausted, new batches of antibody must be produced by regenerating a similar immune response which, even with appropriate validation, often results in subsequent batches displaying variations in antibody performance. mAbs exhibit precise and reproducible binding properties from batch to batch, but the time and cost to generate a mAb is considerably greater compared with pAbs. rAbs offer the specificity and reproducibility of a hybridoma-derived mAb, but do not suffer from clonal drift or gene deletion, which may compromise mAb performance. However, rAbs are costly to produce, are not commercially available for a wide range of target proteins, and may present different production challenges.
Much has been written within the last decade concerning the issue of data reproducibility, especially as it pertains to antibodies. While some commentaries focused on methods, cell lines, critical reagents and data reporting [1,2], others focused on perceived systemic pitfalls of antibody technology in general as the primary culprit [3,4]. Clearly there is a need to better understand the relationship between how antibodies are made and used and how standardized antibody validation strategies could improve the overall performance of antibodies as critical reagents [5–8]. While the majority of commercially produced antibodies are used appropriately in context by researchers resulting in the collection of high-quality and highly reproducible data, some well-validated antibodies are used by researchers inappropriately, for instance in the wrong application, resulting in irreproducible data, and some companies produce antibodies that should not be in the marketplace at all [7]. As the potential economic impact of low reproducibility rates in life science research were assessed [9], thought leaders and key stakeholders assembled to begin to formulate approaches to resolve the dilemma, first by proposing ‘pillars’ for application-specific antibody validation [10] and subsequently by establishing a conceptual framework for validation [11]. The latter meeting correctly identified the many factors contributing to the problem of data irreproducibility (e.g., cell culture methods and cell line variances used for validation being a main focus), and indirectly refuted the premise that the primary cause of the problem was due to “some manufacturers producing consistently good antibodies, and others consistently poor ones” [3] or that the inherent properties of antibodies, such as unwanted cross-reactivity or unacceptably high levels of lot-to-lot variation, were to blame [4], as others initially asserted. With the goal to discuss and improve reproducibility, a diverse group of stakeholders including antibody producers, researchers, teaching institutions, journals and funding agencies needs to act with shared responsibilities. Here we specifically address the notion that “pAbs should be phased out of research entirely” [3]. The adage encouraging us to “use the right tool for the job” presupposes that we have more than one tool in our toolbox. If research is limited to using only one tool, science may fall prey to Maslow’s hammer: “If the only tool you have is a hammer you tend to see every problem as a nail” [12]. We see pAbs as invaluable research tools that continue to serve an appropriate role in research, especially if: (1) they are developed, validated and ultimately manufactured with the correct specifications; (2) positive and negative controls are used appropriately during production and release; and (3) they are ultimately used in an appropriate assay-specific context, according to conditions established for use by the manufacturer. While the inclusion of appropriate positive and negative controls is a requirement when using any form of antibody in all immunoassays, inclusion of controls in experimental designs is essential when using pAbs to assure sensitivity, specificity and reproducibility of results. Acceptable controls may include the use of: (1) purified native or recombinant proteins; (2) cells ectopically expressing the antibody’s target; (3) native cells or tissue endogenously expressing the antibody’s target; (4) cells transfected or engineered to downregulate or eliminate expression of the target; (5) cells stimulated or repressed with a drug, growth factor, an environmental condition or other additive affecting the expression of the antibody’s target; and (6) the inclusion of a positive control or gold-standard antibody run in parallel experiments. These methods are each informative and have differential value. Changes in the genetic code typically causing a complete ‘knockout’ of gene function can be considered a true negative control. This is true for lysate-based assays and cell or tissue staining where correct protein localization is critical. However, in the case of antibodies against post-translationally modified sites (phosphorylation, acetylation, etc.) downregulation of gene expression is not informative because it does nothing to indicate, for instance, phospho-specificity of the antibody. Instead, stimulation or inhibition of a signaling pathway known to modulate a specific site is preferred.
The benefits of clonal & biophysical diversityThe core benefits of pAbs center on two inherent properties: clonal and biophysical diversity. The ‘poly’ clonality of pAbs allows the binding of multiple antigenic determinants of the target. This allows pAbs to be more sensitive in certain assays against a variety of target proteins, cells or organisms. The biophysical diversity of pAbs allows for greater stability when environmental challenges may cause inactivation, lability or precipitation of other forms of antibodies. These two properties are the basis of numerous advantages pAbs offer relative to their mAb and rAb counterparts.
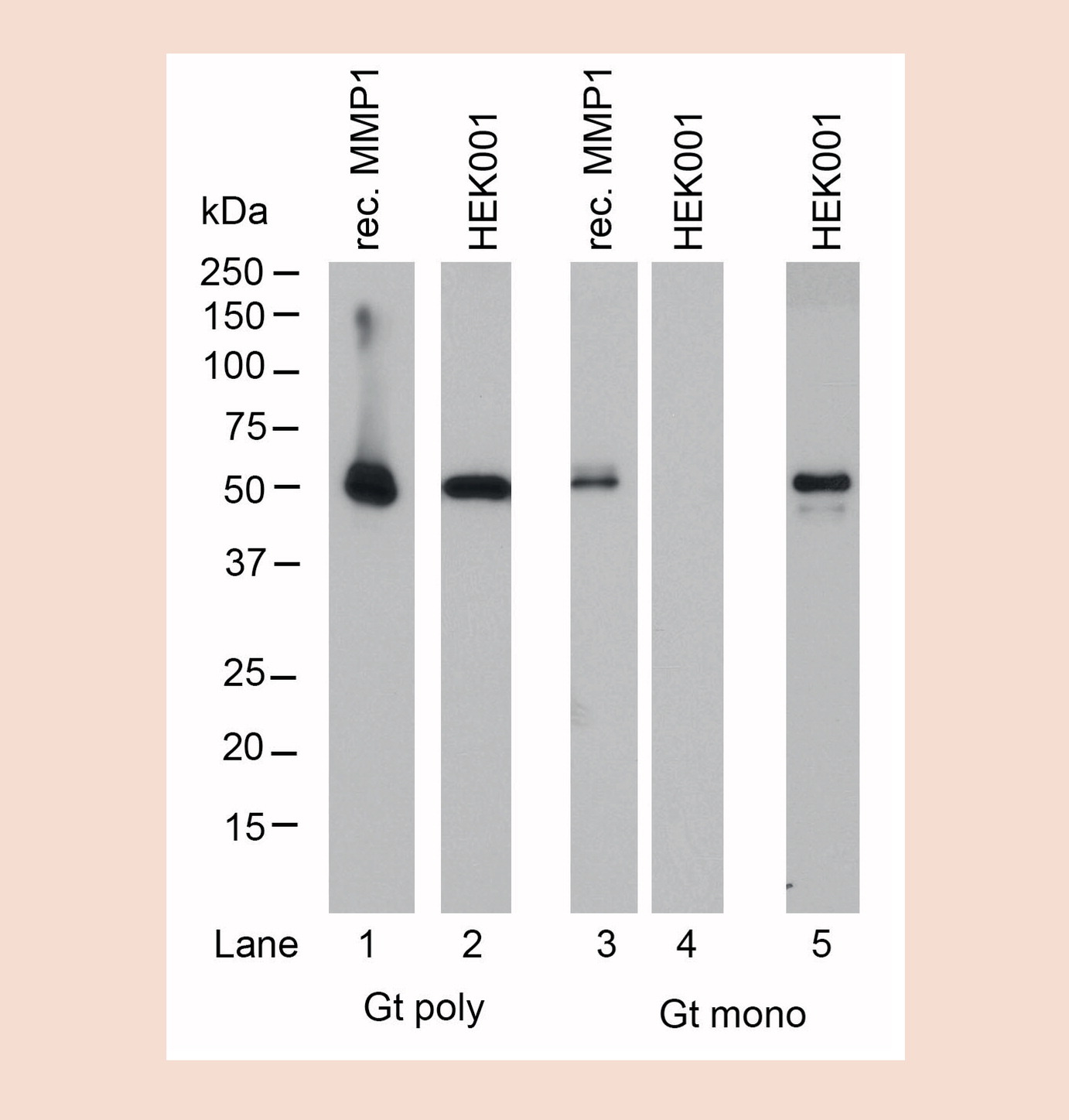
Figure 1. Comparison of sensitivity levels of monoclonal and polyclonal antibodies against MMP1.
In the research environment multi-epitope binding properties for pAbs have clear benefits. The heterogeneous binding of several different epitopes and/or antigens by pAbs renders these reagents more likely to successfully bind a specific antigen in a variety of different test conditions and immunoassays [13], making these tools more appropriate for use than their counter-parts. As capture antibodies in sandwich assays like capture ELISA, pAbs offer higher sensitivity ranges in general than mAb–mAb pairings, due to more effective capturing of multiple antigen variants or epitopes presented by the analytes [14]. This is especially crucial when considering human donor diversity and the need to cover a broad set of naturally occurring variances. Because pAbs typically recognize multiple epitopes on a target protein, they are more effective at detecting a target for use in chromatin immunoprecipitation (ChIP), even if a few epitopes are masked by cross-linking [15]. In immunohistochemistry (IHC) where the effects of the tissue fixation and processing on the epitope is unknown and highly variable, pAbs can be a better option because their multi-epitope binding allows for antigen recognition even if some of these epitopes are affected by changes in an antigen’s tertiary structure or accessibility. mAbs in comparison may find their epitope buried [16]. Furthermore, it has been reported for IHC that pAbs offer greater sensitivity for detecting proteins that are present in low quantities in a sample, since multiple antibodies will bind to multiple epitopes on the protein [17]. A western blotting experiment comparing pAb and mAb derived from the same goat as the original source of antibody show very robust and more sensitive detection by the pAb when compared with its mAbs counterpart (see Figure 1) due to multi-epitope binding. This feature also supports pAbs as an excellent choice to generate secondary antibody reagents since they are less discriminate in their binding sequence and more tolerant of structural variations on primary antibodies. This is especially true when detecting murine mAbs, which have significant isoform variations in sequence, structure and processing and is also crucial when detecting human antibodies in diagnostic assays with patients from varied ethnic backgrounds to prevent or minimize false negatives.
In diagnostic and therapeutic applications, pAbs also demonstrate advantages due to their multi-epitope binding properties. For instance, pAbs are more likely to result in high-avidity binding, the low likelihood of antigen ‘escape variants’ emerging, and the efficient triggering of effector functions [18,19]. pAbs have been shown to eliminate pathogens more effectively than mAbs due to the pAbs ability to bind different structures on a given pathogen, thereby increasing the likelihood of its elimination. Furthermore, mAbs do not represent the benefits of polyclonality utilized by a natural immune system and can therefore be less efficacious as antibody drugs against infectious agents [20]. For this reason, it has been suggested that pAbs may represent an overlooked approach to viral and toxin neutralization in antibody-based therapeutics [21]. In diagnostic assays that are dependent on immune complex formation such as immunoturbidimetric assays, for example, turbidimetry and nephelometry, pAbs are preferred as they can form lattices with homogeneous, monomeric protein antigens because each antibody can interact with a different epitope on the antigen [22]. pAbs represent an ‘elegant solution’ to the problem of detecting contaminating host cell proteins as impurities in biological drugs [23]. pAb development allows for the simultaneous detection of both low and high abundancy proteins and low and high immunogenic proteins in bioprocessing streams [24,25]. Developing monoclonal reagents towards these types of targets would pose significant technical hurdles and in the case of host cell protein detection are currently not feasible or practical.
The biophysical diversity of pAbs allows for generally easier storage and dilution than mAbs, due to their variance in biophysical attributes, such as charge and hydrophobicity. By contrast mAbs may require the addition of stabilizing agents at low concentrations to prevent aggregation, precipitation and preserve antibody binding [26]. Similarly, pAbs are more resistant to changes in temperature and pH when compared with mAbs, which tend to require stabilizers or excipients to maintain their biological activity when exposed to environmental stresses [27]. While it has been reported that pAbs are much easier to couple with small-molecule labels because they are more tolerant of accepting a conjugation moiety without negatively affecting the ability of the antibody to bind the target, this is most likely due to selective coupling of subspecies within the polyclonal mixture. For similar reasons, mAbs have been shown to demonstrate narrower ranges of the degree-of labeling (DOL) to optimize signal intensity [28].
Additional benefits of using pAbsIn addition to the benefits conferred on pAbs resulting from their clonal and biophysical properties, other benefits can be ascribed to pAbs due to methods-based production differences in comparison with mAbs. pAbs can be generated in a wide range of host animals including avian, which have been reported to elicit a stronger immune response to proteins that have high homologies between mammalian targets and mammalian hosts [29]. Furthermore, pAb generation provides the researcher with opportunities to modify the quantity of the antigen, the route of injection, the number and distribution of injection sites, the frequency of antigen injection, the particular adjuvant, the quantity of the adjuvant and the antigen:adjuvant ratio in a manner that may not be typically available when conventional mAbs are produced. When optimized, these parameters may result in higher affinity antibodies with increased yields for pAb production [30]. Especially when goats are used as the animal of choice, vastly greater amounts of pAbs can be generated within 3–4 months in general, at a lower cost, and with less technical skill than is required to produce mAbs or rAbs [31]. Lastly, pAbs have fewer facility and specialized equipment requirements; for instance, pAbs do not require cell culture facilities, devices for electroporation or advanced skills for cloning.
Factors influencing the decision to use pAbspAbs can be produced in a wide variety of host species, considerably greater in number and diversity than mAbs, including conventional hosts such as rabbits, goats and sheep, as well as specialized hosts like chicken, duck [32] and donkey. Consequently, there are several factors that need to be considered when deciding on: (1) what type of antibody to make (e.g., pAbs, mAbs or rAbs); and (2) if pAbs are the desired type, which host species to use. If the pAb is intended to be used in an application requiring large amounts of antibody over time, a host with greater body weight, such as donkey, goat or sheep, and therefore greater total blood volume may be the better choice, due to the increased yield of serum as well as the longevity of the animal. Mouse pAbs, while a cheap and fast way of producing an antibody serum, are not easily maintained or reproduced and should be avoided for these reasons. A poor response to mouse immunogens may be observed when mice or rats are immunized for pAb or mAb development. In some instances, immunizing a heterologous species such as hamster may result in a stronger immune response [33].
Strategies to minimize the disadvantages inherent to pAbspAbs do have disadvantages mainly due to their finite supply. However, many of these disadvantages, when properly addressed, can be sufficiently mitigated to allow for the collection of high-quality data that can be reproduced from experiment to experiment and from laboratory to laboratory. For example, pAbs may require more rigorous validation than mAbs due to their heterogeneity. Many pAbs are raised against native proteins or fragments of proteins, rather than against peptides, as is commonly done when mAbs are produced. Therefore, a thorough analysis of the homology of the immunogen used with other proteins is crucial. Since pAbs will recognize multiple epitopes, the risk of cross-reactivity with different targets is inherent. Some of those risks can be eliminated by negative absorption during affinity purification on a column containing the closely related protein. Many immunogens are conjugated to a carrier protein such as KLH, BSA or OVA, that will also elicit an unwanted immune response when co-immunized into the host with the target protein. Again, this cross-reactivity can be eliminated during the serum purification steps by cross adsorption using a carrier-specific column or by avoiding carriers altogether during immunization, as many proteins do not require carriers as previously thought due to their inherent structure and size compared with haptens and peptides. Cross absorption is especially useful for secondary reagents to remove unwanted species or isotype cross-reactivities. After purification, the specificity of pAbs must be ensured for every lot produced, for instance by testing the antibody against a panel of closely related and control recombinant proteins by direct ELISA or western blot (WB). Since pAbs often are collected from antiserum generated over a period of time (sometimes many years), lot-to-lot consistency must be controlled. This can be achieved using methods similar to those used for mAb production, where each newly purified lot is tested and the results compared to its original specifications and applications under the same experimental conditions to assess for drift in lot-to-lot reproducibility. Lot-to-lot consistency can be effectively managed by comparing the performance of newly manufactured lots to existing and historical lots. Pooling is another risk mitigation strategy as it has the intended effect of both minimizing variations in antibody reactivity and results in fewer, yet larger, lots produced. Very large pools of serum (usually obtained from donkey, sheep or goat) are easier to maintain over many years than multiple smaller lots produced from hosts that yield less serum and therefore greater lot-to-lot consistency can be achieved. If the pAb is carefully developed and production parameters are carefully maintained, the end product is just as valuable and reliable a reagent as a mAb. By example, the specificity for pAbs can be shown utilizing CRISPR knock-out cell lines for validation (Figure 2A & B) as effectively as genetic modifications can be used to demonstrate specificity for mAbs.
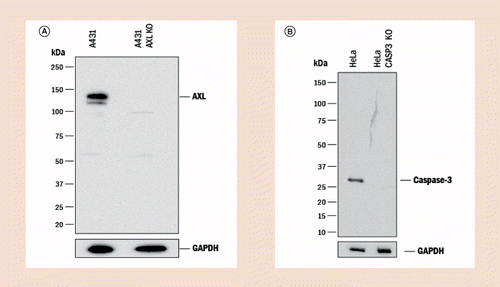
Figure 2. Knockout data showing specificity of polyclonal antibodies against Caspase 3 and Axl.
While pAbs are an invaluable research tool for many applications and fit specific needs of certain immunoassays, they clearly are not the best choice in other areas especially where regulatory controls exist. For instance, epitopes on targets that represent a small change such as a single amino acid polymorphism [34,35] or a post-translational modification [36,37] (e.g., phosphorylation, methylation, hydroxylation), may be better targeted using a mAb. Often these types of targets or changes are too small to elicit a robust pAb immune response and in addition increase the risk of nonspecific antibody reactivity within the pAb mixture. Despite this disadvantage, pAbs can be generated successfully against post-translational modifications (PTMs) or SNPs by immunoaffinity purification of high titer antiserum and by utilizing the additional step(s) of negative adsorption as stated above. However, attention to optimized protocols and the inclusion of adequate controls and standards is essential to achieve lot-to-lot reproducibility and maintain acceptable binding affinities. For similar reasons epitopes for isozyme- [38] or idiotype- [39] specific targets may be better recognized by mAbs to ensure the highest specificity possible. For diagnostic or therapeutic uses additional factors that can negatively influence the decision of choosing a pAb over a mAb or a rAb include the regulatory status of the final product. If the antibody is used in a diagnostic application such as IHC, the requirements of regulatory agencies such as the US FDA and its counterparts elsewhere must be considered from the onset of antibody production. For instance, antibodies used in an approved diagnostic test may require the disclosure or knowledge of the epitope the antibody recognizes on the target of interest [40]. This analysis would be difficult, if possible at all, using a pAb. Another factor to consider for diagnostic use would be a stable supply of material, without changes in the sensitivity and specificity of the antibody. Since pAbs are inherently more prone to changes over the duration of the immunization cycle, variations in performance of concern to regulatory agencies can be minimized or eliminated by choosing a mAb or rAb. Although pAbs are well suited for certain therapeutic applications as stated above, mAbs or rAbs would be a more desirable alternative when considering the potential for patients to develop anti-drug antibodies, as a single antibody, in contrast to a mixture of pAbs, would be easier to analyze for interference with clinical efficacy [41]. Similarly, mAbs have a long history of regulatory approval for therapeutics including for use as antibody-drug conjugates [42], as vaccines to rapidly emerging infectious diseases [43], to target multifactorial diseases [44], for antibody-dependent cell-mediated cytotoxicity [45], and many other clinical applications previously reviewed[46–49].
ConclusionpAbs, mAbs and rAbs represent a collection of invaluable tools for life science research and each form of antibody has advantages and disadvantages when compared with their counterparts. These differences can be exploited to create opportunities for the collection of high-quality reproducible data in an application-specific setting. What is clear is that antibodies, including pAbs, have been, are now, and will continue to be critical reagents most often used by life science researchers, and that antibodies are transformative tools used to diagnose and treat disease. The life science research community needs to better understand the strengths and weaknesses of each of these tools and not to dismiss an entire class of antibodies due to perceived limitations. Each type of antibody should be deployed in a context that appropriately takes advantage of their inherent properties. For some researchers, pAbs may represent an overlooked tool that cannot easily be replaced by using a mAb or rAb. By emphasizing the appropriate role pAbs may play in conducting life science research our efforts here should improve upon the use of antibodies, the collection of reproducible data and the expansion of antibody-based technologies. This is especially important as the demands of platform technologies continue to grow, new targets of interest are discovered and rarely studied species are explored.
Future perspectiveMarket pressure, rather than regulatory oversight, will effect changes in how antibodies, including pAbs, are produced and validated. These changes will likely have the greatest impact on a subset of producers who have not yet adopted high standards for antibody production and validation. The life science research community would greatly benefit from universal standards for antibody validation that are application-specific in their approach to validation. The perspective of researchers will likely evolve, resulting in a better understanding of when, and when not, to use an antibody in a ‘fit-for-purpose’ manner. Closing this knowledge gap may result in the appropriate deployment of pAb, mAb or rAb forms of antibodies for the collection of high-quality data. As funding agencies and some journals have already started to modify requirements to enhance the specification of critical reagents in grants and publications, it is likely universities and antibody producers themselves will bolster efforts to better educate researchers on antibody selection and use in an application-specific context. While pAbs will likely always play an appropriate role as a research tool as described in this review, the role for rAbs will likely expand as this technology continues to develop.
Author contributionsCAA and BA contributed equally to preparing this manuscript.
Financial & competing interests disclosureCAA is an employee of Rockland Immuno-chemicals Inc., a company that produces and sells antibodies. BA is an employee of Bio-Techne, a company that produces and sells antibodies. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Open accessThis work is licensed under the Creative Commons Attribution 4.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
ReferencesPapers of special note have been highlighted as:
• of interest; •• of considerable interest
- Begley CG, Ellis LE. Raise standards for preclinical cancer research. Nature 483, 531–533 (2012). •• Identifies the quality of preclinical data as a significant factor when confirming clinical studies.
- Francia G, Kerbel RS. Raising the bar for cancer therapy models. Nat. Biotech. 28(6), 561–562 (2010).
- Bradbury A, Plückthun A. Standardize antibodies used in research. Nature 518, 27–29 (2015). •• Calls for the phasing out of polyclonal antibodies from research entirely.
- Baker M. Blame it on the antibodies. Nature 521, 274–276 (2015).
- Bordeaux J, Welsh AW, Agarwal S et al. Antibody validation. Biotechniques 48(3), 197–209 (2010). • Calls for universally accepted guidelines for antibody validation as a means of showing antibody specificity, selectivity and reproducibility.
- Weller MG. Quality issues of research antibodies. Anal. Chem. Insights 11, 21–27 (2016). • Comprehensive analysis of irreproducibility that addresses the complexity of the problem.
- Ascoli CA, Birabaharan J. The antibody dilemma: shortcuts taken by antibody manufacturers and end-users have led to a reproducibility crisis. Genetic Engin. Biotechnol. 35(14), (2015). • Describes a knowledge gap between how antibodies are produced and intended for use.
- Polakiewicz RD. Antibodies: the solution is validation. Nature 518, 483 (2015).
- Freedman LP, Cockburn IM, Simcoe TS. The economics of reproducibility in preclinical research. PLoS Biol. 13(6), e1002165 (2015).
- Uhlen M, Bandrowski A, Carr S et al. A proposal for antibody validation. Nat. Methods 13(10), 823–827 (2016). •• Establishes immunoassay-specific conceptual pillars for antibody validation.
- Antibody Validation: Standards, Policies, and Practices. September 25–27, 2016, Asilomar Conference Grounds, Pacific Grove California. Organized by the Global Biological Standards Institute and The Antibody Society. www.gbsi.org/event/antibody-validation-2016/
- Maslow AH. The Psychology of Science: A Reconnaissance. Harper & Row, NY, USA. (1966).
- Stills HF. The Laboratory Rabbit, Guinea Pig, Hamster, and Other Rodents. Chapter 11. In: Polyclonal Antibody Production. Suckow MA, Stevens KA, Wilson RP (Eds). Academic Press, MA, USA, 259–270 (2011).
- Cox KL, Devanarayan V, Kriauciunas A, Manetta J, Montrose C, Sittampalam S. Assay Guidance Manual [Internet]. In: Immunoassay Methods. Sittampalam GS, Coussens NP, Brimacombe K et al. (Eds). Eli Lilly & Company and the National Center for Advancing Translational Sciences, MD, USA, 223–266 (2012). www.ncbi.nlm.nih.gov/books/NBK92434
- Guide to chromatin immunoprecipitation: critical factors for success. Technical Guide. Literature # TP5994EN00. EMD Millipore Corporation. MA, USA (2012).
- Helsby M. Immunohistochemistry: monoclonal or polyclonal? CiteAb/Blog (2013). https://blog.citeab.com/immunohistochemistry-monoclonal-or-polyclonal/
- Ivell R, Teerds K, Hoffman GE. Proper application of antibodies for immunohistochemical detection: antibody crimes and how to prevent them. Endocrinology 155(3), 676–687 (2014).
- Sharon J, Sompuram SR, Yang CY, Williams BR, Sarantopoulos S. Construction of polyclonal antibody libraries using phage display. In: Antibody Phage Display. Methods in Molecular Biology. Walker JM (Ed.). Humana Press, NY, USA, 101–112 (2002).
- Sharon J, Sarantopoulos S, Den W et al. Recombinant polyclonal antibody libraries. Comb. Chem. High Throughput Screening 3(3), 185–196 (2000).
- Bregenholt S, Jensen A, Lantto L, Hyldig S, Haurum JS. Recombinant human polyclonal antibodies: a new class of therapeutic antibodies against viral infections. Curr. Pharm. Des. 12(16), 2007–2015 (2006).
- Newcombe C, Newcombe AR. Antibody production: polyclonal-derived biotherapeutics. J. Chromatography B 848(1), 2–7 (2007).
- Blirup-Jensen S. Protein standardization III: method optimization. Basic principles for quantitative determination of human serum proteins on automated instruments based on turbidimetry or nephelometry. Clin. Chem. Lab. Med. 39(11), 1098–1109 (2005).
- Bracewell DG, Francis R, Smales CM. The future of host cell protein (HCP) identification during process development and manufacturing linked to a risk-based management for their control. Biotechnol. Bioeng. 112(9), 1727–1737 (2015).
- Chimento DP, Abarca KA. Developing antibodies for detection of host cell protein contaminants. bioperspectives. Genengnews.com (2014). www.genengnews.com/bioperspectives/developing-antibodies-for-detection-of-host-cell-protein-contaminants/5118
- Rusbuldt J, Abarca-Heidemann K, Chimento D, Ascoli C. Effectiveness of 2-D electrophoresis over 1-D electrophoresis followed by western blotting for evaluating strategies used to generate host cell proteins antibodies. Bulletin 6405, Tech Note Bio-Rad Laboratories, CA, USA (2013). www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6405.pdf
- Kueltzo LA, Wang W, Randolph TW, Carpenter JF. Effects of solution conditions, processing parameters, and container materials on aggregation of a monoclonal antibody during freeze–thawing. J. Pharm. Sci. 97(5), 1801–1812 (2008).
- Kheddo P, Tracka M, Armer J et al. The effect of arginine glutamate on the stability of monoclonal antibodies in solution. Int. J. Pharm. 473(1–2), 126–133 (2014).
- Vira S, Mekhedov E, Humphrey G, Blank PS. Fluorescent labeled antibodies – balancing functionality and degree of labeling. Anal. Biochem. 402(2), 146–150 (2010).
- Tini M, Jewell UR, Camenisch G, Chilov D, Gassmann M. Generation and application of chicken egg-yolk antibodies. Comp. Biochem. Physiol. 113(3), 569–574 (2002).
- Carey Hanly W, Artwohl JE, Taylor Bennett B. Review of polyclonal antibody production procedures in mammals and poultry. ILAR J. 37(3), 93–118 (1995).
- Lipman NS, Jackson LR, Trudel LJ, Weis-Garcia F. Monoclonal versus polyclonal antibodies: distinguishing characteristics, applications, and information resources. ILAR J. 46(3), 258–268 (2005).
- Chiou VY. The development of IgY(DeltaFc) antibody-based neurotoxin antivenoms and the study on their neutralization efficacies. Clin. Toxicol. 46(6), 539–544 (2008).
- Kascsak RJ, Rubenstein R, Merz PA et al. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J. Virol. 61(12), 3688–3693 (1987).
- Rogers LM, Mott SL, Smith BJ, Link BK, Sahin D, Weiner GJ. Complement-regulatory proteins CFHR1 and CFHR3 and patient response to anti-CD20 monoclonal antibody therapy. Clin. Cancer Res. 23(4), 954–961 (2017).
- Shimizu M, Matsuda A, Yanagisawa K et al. Functional SNPs in the distal promoter of the ST2 gene are associated with atopic dermatitis. Hum. Mol. Gen. 14(19), 2919–2927 (2005).
- Zhang L, Michelle English A, Bai DL et al. Analysis of monoclonal antibody sequence and post-translational modifications by time-controlled proteolysis and tandem mass spectrometry. Mol. Cell Proteomics 15, 1479–1488 (2015).
- Lyon K, Stasevich TJ. Imaging translational and post-translational gene regulatory dynamics in living cells with antibody-based probes. Trends Genet. 33(5), 322–335 (2017).
- Hu X, Cheng S, Liu X et al. Development of monoclonal antibodies and immunochromatographic lateral flow device for rapid test of alanine aminotransferase isoenzyme 1. Protein Exp. Purif. 119, 94–101 (2016).
- Muller S, Parsons MS, Kohler H, Grant M. The significance of a common idiotype (1F7) on antibodies against human immune deficiency virus Type 1 and hepatitis C virus. Front. Oncol. 6(11), 1–7 (2016).
- US FDA. Guidance for Submission of Immunohistochemistry Applications to the FDA. 2017. Title 21 – Food and Drugs. Chapter I – Food and Drug Administration, Department of Health and Human Services, Subchapter H – Medical Devices, Part 864 – Hematology And Pathology Devices, Subpart B – Biological Stains, Sec. 864.1860 Immunohistochemistry reagents and kits. www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=864.1860
- Darrouzain F, Bian S, Desvignes C et al. Immunoassays for measuring serum concentrations of monoclonal antibodies and anti-biopharmaceutical antibodies in patients. Ther. Drug. Monit. 39(4), 316–321 (2017).
- Shepard HM, Phillips GL, Thanos C, Feldmann M. Developments in therapy with monoclonal antibodies and related proteins. Clin. Med. (Lond.) 17(3), 220–232 (2017).
- Wang Q, Yan J, Fu Gao G. Monoclonal antibodies against Zika virus: therapeutics and their implications for vaccine design. J. Virol. 9, e01049–17 (2017).
- Grainge CL, Maltby S, Gibson PG, Wark PAB, McDonald VM. Targeted therapeutics for severe refractory asthma: monoclonal antibodies. Expert Rev. Clin. Pharmacol. 9(7), 927–941 (2016).
- Carvalho S, Levi-Schaffer F, Sela M, Yarden Y. Immunotherapy of cancer: from monoclonal to oligoclonal cocktails of anti-cancer antibodies: IUPHAR Review 18. Br. J. Pharmacology 173(9), 1407–1424 (2016).
- Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 15(6), 361– 370 (2015).
- Liu JKH. The history of monoclonal antibody development – progress, remaining challenges and future innovations. Ann. Med. Surg. (Lond.) 3(4), 113–116 (2014).
- Glassman PM, Balthasar JP. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol. Med. 11(1), 20–23 (2014).
- Gharwan H, Groninger H. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nat. Rev. Clin. Oncol. 13(4), 209–227 (2015).
First draft submitted: 10 May 2018; Accepted for publication: 21 June 2018
Address correspondence to: Carl A Ascol, Rockland Immunochemicals Inc., Limerick, PA, USA; E-mail: carl.ascoli@rockland-inc.com
To purchase reprints of this article contact: s.cavana@future-science.com
Copyright © 2018 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


