



July 6, 2021 -- A new single-cell sequencing method yields at least 10 times more data for each individual cell compared to previous techniques. The method, published in Nature Biotechnology on July 5, may help cancer researchers uncover new insights into the biological underpinnings of cancer such as drug resistance.
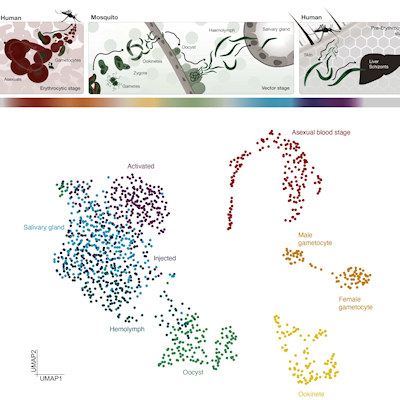
Single-cell genomics is a transformative platform that enables researchers to distinguish properties of individual cells in a given tissue. This can be an especially powerful tool for cancer research, since the cells of a tumor are strikingly diverse. As a tumor grows, it acquires various mutations, and changes in gene activity give rise to subpopulations of tumor cells with new qualities, such as the ability to spread to other body parts or resist anticancer drugs.
Despite its potential, single-cell research is currently hampered by limitations in the number of cells that can be analyzed at this depth of detail.
To address this limitation, researchers from Oregon Health and Science University (OHSU) previously developed a strategy called single-cell combinatorial indexing (sci), which uses a transposase-based library construction to assess a variety of genomic properties in a high-throughput manner. While the technique greatly increases the number of single cells that can be analyzed in one experiment, it comes with the tradeoff of sparse coverage of readable DNA sequences per cell.
The technique relies on the sequencing of DNA libraries -- collections of DNA fragments that can be used to analyze genes and mutations for thousands of single cells at the same time. The process uses an enzymatic reaction to attach primers (adapters) to both ends of each DNA fragment. Many sci methods only achieve this around 50% of the time.
The team took the method a step further, improving the number of usable reads obtained per cells by utilizing an adapter-switching strategy called "s3." This provides an approximately tenfold improvement in the amount of DNA that can be recovered from a single cell to be sequenced and interpreted.
"The downside has been the low quality of single-cell profiles," said senior author Andrew Adey, PhD, associate professor of molecular and medical genetics in the OHSU School of Medicine, in a statement. "Our method allows us to get a much more complete profile of any given single cell."
Adapter replacement is used to produce DNA fragments tagged with both forward and reverse adapters. The team uses multiple rounds of extension before polymerase chain reaction (PCR) to achieve this yield improvement.
"They are kind of like handles the sequencer uses to start reading," Adey explained.
First, a Tn5 mosaic end sequence (primer for transposase) is incorporated into the forward primer sequence to create a reaction-specific DNA barcode. The use of a uracil-intolerant polymerase prevents extension beyond the end of the DNA fragment. Then, a second template oligo is introduced, which anneals the copied mosaic end sequence to extend the molecule with the incorporation of a reverse adapter. This process results in library fragments that have both forward and reverse adapter sequences.
Improving single-cell techniques for cancer research
Adey and colleagues showed that their method can be used to reveal DNA alterations that have emerged in a subset of cells in tumor samples taken from patients with pancreatic cancer. By conducting whole-exome sequencing of pancreatic cancer cell lines derived from patient tumors, the team was able to detect unique subpopulations of cells with copy number loss of certain tumor suppressor genes such as cyclin-dependent kinase inhibitor 2A (CDKN2A), SMAD family member 4 (SMAD4), and BRCA2. They also identified in the pancreatic ductal adenocarcinoma-2 line, a subclonal amplification of serine protease 1 (PRSS1), a mutation associated with tumor size and tumor node metastasis rate.
"You can potentially identify rare cell subtypes within a tumor that are resistant to therapy," Adey said.
This knowledge could be used to develop individualized treatment plans for patients.
"If you are working with something like a cancer biopsy from a patient, a very small piece of tissue, you really want to make every cell count, you really have to get a lot of info from each single cell," Adey added.
In addition to improving the efficiency of the single-cell sequencing process, the method also has the added benefit of reducing sequencing costs by about one-third, according to the team. The platform utilizes adapters that are designed for standard sequencing techniques, instead of requiring custom primers or workflows in other competing methods. This makes the s3 platform compatible with many other single-cell workflows.
"This chemistry can just slot into many other assays," Adey said. "People can just start using it."
Do you have a unique perspective on your research related to cell biology or genomics? Contact the editor today to learn more.
---


Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


