



April 5, 2021 -- A new large-scale study indicates x-ray crystallography can be used to find drugs that could be repurposed to target the SARS-CoV-2 main protease. In addition to identifying 37 potential drug candidates, the study, published in Science on April 2, revealed a new binding site on the SARS-CoV-2 main protease to which drugs can bind.
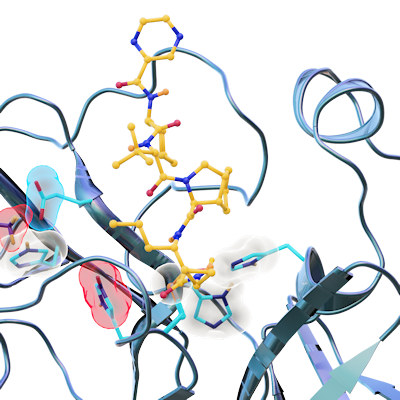
Proteolytic cleavage of the SARS-CoV-2 polyprotein is primarily accomplished by the main protease. The enzyme contains a chymotrypsin-like fold appended with a C-terminal helical domain, and it harbors a catalytic dyad comprised of Cys145 and His41 in its active site, which is formed by four major pockets. The active site is located in a cleft between the two N-terminal domains of the three-domain structure of the monomer, while the C-terminal helical domain is involved in regulation and dimerization of the enzyme.
The main protease is a target for antiviral drug discovery and compound screening because of its involvement in virus replication. In the new study, researchers from the German Electron Synchrotron (DESY), a national research center in Germany that operates particle accelerators used for structural biology experiments, sought to identify repurposed drug candidates that might be effective against the main protease.
The DESY team conducted high-throughput x-ray crystallography screens of two drug libraries to find candidates that could be used against the SARS-CoV-2 main protease. The P11 high-throughput crystallography beamline at PETRA III (DESY's high-brilliance x-ray light source facility) was used to image cocrystals of the main protease mixed with compounds from the drug libraries at atomic resolution.
In all, 5,953 unique compounds were screened from the Fraunhofer IME Repurposing Collection of the Fraunhofer Institute for Translational Medicine and Pharmacology and the "Safe-in-man" library from the Italian company Dompé Farmaceutici. In contrast to many crystallographic fragment-screening experiments, these drug libraries include whole, chemically complex compounds. The compounds have been tested for bioactivity and cell-permeability in clinical trials.
The PETRA III station P11 includes fully automated sample changes with a robotic arm, so that each of the more than 7,000 measurements took only about three minutes. Using this high-throughput technique, the team was quickly able to separate compounds of interest from those with no potential activity.

The sample holder (left) at measuring station P11 rotates the sample in the x-ray light from the PETRA III accelerator. Meanwhile, a stream of cold nitrogen cools the sample. With the help of many scattering images from different directions, the molecular structure of the protein-drug complex can be calculated. Image courtesy of DESY/C/ Schmid.
The researchers obtained a total of 2,381 unique compounds from the x-ray diffraction datasets, which were then subjected to automated structure refinement, cluster analysis, and pan-dataset density analysis (PanDDA). From the 3D protein structures, they identified 37 compounds that bind to the main protease.
"A particular strength of our method of x-ray screening compared to other screening methods is that we obtain the three-dimensional structure of the protein-drug complexes as a result and can thus identify the binding of the drugs to the protein at the atomic level," explained co-author Patrick Reinke, PhD, DESY postdoctoral researcher. "Even if the two most promising candidates do not make it into clinical trials, the 37 substances that bind to the main protease form a valuable database for drug developments based on them."
Most of the compounds were bound in the active site of the main protease. Eight bind covalently and an additional eight bind noncovalently within the active site. Thirteen of the compounds bind outside the active site in various locations.
To further explore the suitability of the compounds, cell-based viral reduction assays were conducted to evaluate antiviral activity. The researchers identified a total of seven compounds -- one peptidomimetic and six nonpeptidic compounds -- that exhibited at least a 100-fold reduction in viral particles with a favorable selective index (ratio that measures the window between cytotoxicity and antiviral activity). The seven compounds were AT7519, calpeptin, ifenprodil, MUT056399, pelitinib, tolperisone, and triglycidyl isocyanurate.
"The active substances Calpeptin and Pelitinib clearly showed the highest antiviral activity with good cell compatibility. Our cooperation partners have therefore already started preclinical investigations with these two substances," explained first author and DESY postdoctoral researcher Sebastian Günther, PhD.
Unexpected revelations
During their analysis of the full drug compounds, the researchers found something unexpected -- a binding site on the main protease that had been completely unknown. This allosteric site was found in the hydrophobic pocket of the C-terminal dimerization domain, which is close to the S1 binding site of the spike protein. The team's screening and cell-based assays showed that pelitinib has the potential for antiviral inhibition through binding at this site. Moreover, the hydrophobic nature of the residues in this binding site are conserved in all human coronavirus main proteases.
"It was not only a nice surprise that we were able to discover a new drug binding site on the main protease -- a result that can really only be achieved at a synchrotron light source like PETRA III -- but that even one of the two promising drug candidates binds precisely to this site," said co-author, Christian Betzel, PhD, from the University of Hamburg.
They also identified a second allosteric site that is formed by the deep groove between the catalytic domains and the dimerization domain. While AT7519, the only compound in the screen that bound to this site, had only moderate activity, the scientists suggest the site may be a new target through modulation of Arg298, a residue crucial for enzyme dimerization.
"The investigations at PETRA III impressively show how relevant high-brilliance synchrotron lightsources are for the development of future medicines and for health research as a whole," stressed Helmut Dosch, PhD, chairman of the DESY Directorate. "We must and want to expand our infrastructures even more in the future to cope with health crises like the current one."
Do you have a unique perspective on your research related to infectious diseases or crystallography? Contact the editor today to learn more.
---

Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


