



March 24, 2021 -- Scientists have used atomic maps of hydrogen atoms to determine that the SARS-CoV-2 main protease acts in unexpected ways when it comes into contact with a drug inhibitor. The research, published in the Journal of Medicinal Chemistry on March 23, provides key insights for efforts to repurpose existing drugs to develop candidates for treating COVID-19.
A vital step in the viral replication cycle is the cleavage of two polyproteins encoded by the viral replicase gene into individual functional viral proteins. Each polyprotein is mainly processed, or hydrolyzed, by a chymotrypsin-like protease or main protease that belongs to the class of cysteine protease enzymes. In the case of SARS-CoV-2, the enzymatic activity of its main protease is essential for the production of infectious virions and is therefore a crucial drug target for small molecule protease inhibitors.
The SARS-CoV-2 main protease is composed of two catalytically active subunits (with three functional domains each) that interact through noncovalent interactions. Its active-site cavity is a shallow cleft located on the protein surface between domains I and II.
Ligand-free and ligand-bound drug targets are essential to steer structure-guided and computer-assisted drug design approaches in the right direction. Traditionally, detailed atomic pictures are generated using x-ray cryo-crystallography. However, with this methodology, hydrogen atoms are usually overlooked because they cannot be located based on the x-ray data.
Knowledge of where hydrogen atoms relocate due to inhibitor binding can provide useful information on how protonation states (the addition or loss of protons to a biomolecule, particularly to hydrogen ions) are changed in the protein active-site cavity, thus improving drug design.
Alternatively, neutron crystallography can be used to determine the positions of hydrogen and its heavier isotope, deuterium (D), in biomacromolecules. A benefit of the method is that neutron diffraction data is collected at room temperature, avoiding possible artifacts produced during cryo-cooling of protein crystals (as with x-ray cryo-crystallography).
Using neutron crystallography to map the SARS-CoV-2 main protease
In previous studies, researchers at the U.S. Department of Energy's Oak Ridge National Laboratory (ORNL) conducted a neutron crystallographic study of the ligand-free SARS-CoV-2 main protease, which provided a direct visualization of hydrogen atom locations and hydrogen bonding interactions throughout the enzyme. From this work, the team was able to locate sites where a protease inhibitor would bind with the enzyme.
In the current study, the researchers used neutron crystallography to discover unforeseen changes in the electric charges in the drug binding site of the SARS-CoV-2 main protease that were not predicted by prevailing computer simulations.
The team reported a room-temperature neutron structure of the SARS-CoV-2 main protease in complex with telaprevir to accompany their previously acquired x-ray crystallography data. Telaprevir is a covalent protease inhibitor that belongs to a class of antiviral agents called peptidomimetic inhibitors or alpha ketoamides, which use strings of unnatural amino acids to bind specific protein targets. It is also a U.S. Food and Drug Administration-approved drug used to treat hepatitis C viral infection.
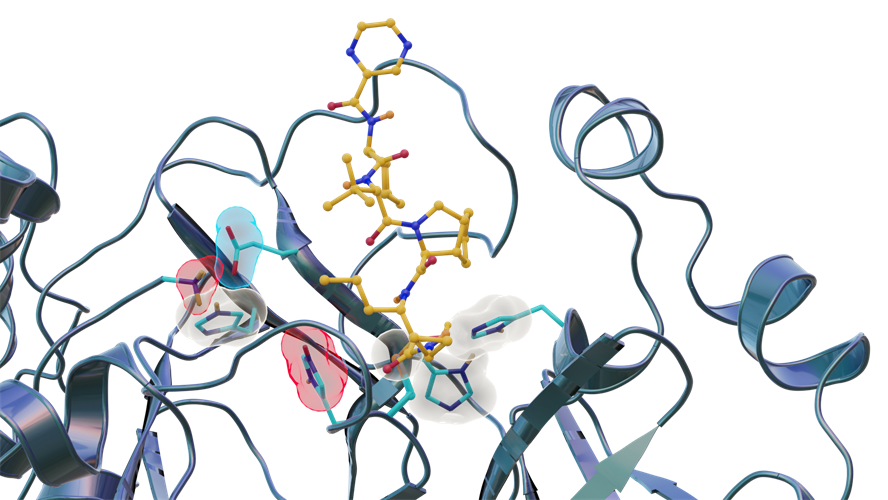
Neutron scattering experiments show electric charges, shown in red, blue and gray, in the SARS-CoV-2 main protease site where telaprevir binds to the structure. The experiments provide critical data for the design of small-molecule drugs to treat COVID-19. Image courtesy of Jill Hemman and Michelle Lehman/U.S. Department of Energy ORNL.
"We found this particular protein -- the SARS-CoV-2 main protease -- which a lot of researchers are studying by computational methods, is behaving in a surprising way," said lead author Daniel Kneller, PhD, a postdoctoral research associate at ORNL, in a statement. "Our findings provide critical experimental data needed to improve computer modeling so that simulations more closely match reality."
The researchers identified three key histidine residues (His41, His163, and His164) in the SARS-CoV-2 main protease active-site cavity are altered after telaprevir binds. Specifically, His163 protonation, driven by inhibitor binding, induces a conformation change leading to stronger hydrogen bonds that may enhance inhibitor binding affinity.
"In this study, we discovered that when telaprevir bound to the main protease, the protonation states [proton addition or loss] in the binding site altered, meaning the locations of the hydrogen atoms changed; essentially, some amino acid sites either gained or lost hydrogen atoms, which changed their electric charges," said author Andrey Kovalevsky, PhD, a macromolecular crystallographer at ORNL. "However, the overall electric charge of the protease inhibitor binding site remained the same as it was before it was attached to the drug molecule. That's something we didn't expect and wasn't correctly predicted in computer simulations."
The researchers noted that assumptions about binding behaviors should not be based purely on the properties of the protease structure before an inhibitor is bound.
"Protein behavior at the level of individual atoms is notoriously difficult to predict. Simulations have to be designed based off ideal scenarios of general chemical knowledge and mathematics, but proteins don't always adhere to ideal scenarios," Kneller said.
The team concluded that the binding of a covalent inhibitor can modulate the protonation states of histidine residues in the active-site cavity of SARS-CoV-2 main protease. In this way, the main protease can tune its electrostatics to accommodate inhibitor and substrate binding into its active-site cleft.
"We believe what we learn from the interactions between telaprevir and the protease enzyme should be transferable to other covalent peptidomimetic inhibitors such as those being investigated right now by major pharmaceutical companies," added author Leighton Coates, Second Target Station instrument systems science and technology manager at ORNL.
Do you have a unique perspective on your research related to virology or cell biology? Contact the editor today to learn more.
---


Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


