



November 3, 2021 -- An interdisciplinary team of engineers and scientists has released DINC-COVID, a web-based portal where researchers can screen COVID-19 drug candidates that might attack specific SARS-CoV-2 proteins. Details of the new platform were published online in Computers in Biology and Medicine.
The DINC-COVID platform performs ensemble docking of small molecules and peptides to SARS-CoV-2, with the goal of screening candidate ligands (reactive molecules) against different conformations of the virus' proteins and their binding pockets. In computational structure-based drug discovery, ensemble docking refers to the generation of an "ensemble" of drug target conformations against which candidate ligands can be docked.
"DINC-COVID offers a ready-to-use solution for researchers to account for protein flexibility while testing compounds against SARS-CoV-2 proteins," wrote the authors, led by Sarah Hall-Swan, a Rice University graduate student and co-lead author of the paper, along with Didier Devaurs, PhD, a research fellow at the University of Edinburgh.
Implementation of DINC-COVID
Hall-Swan and Devaurs developed the portal at the laboratory of co-author Lydia Kavraki, PhD, a computer scientist at the George R. Brown School of Engineering at Rice University.
DINC, which stands for docking INCrementally, is a protocol developed by Kavraki's lab in 2013 to speed protein-peptide docking simulations using parallelization. To take advantage of the parallel architecture of the DINC protocol, the web server is hosted on a 16-core virtual machine, which allows DINC-COVID to run currently on 16 parallel threads (Comput Biol Med, October 15, 2021, Vol. 139).
On the DINC-COVID web server, users choose a binding target, select one of the available ensembles, and upload a ligand of interest. DINC-COVID then performs the ensemble docking using the DINC algorithm. Once all the protein-ligand binding modes are generated, they are rescored and ranked using three scoring functions: AutoDock Vina, AutoDock 4, and Vinardo. Finally, the top-scoring binding modes for each scoring function are returned to the user.
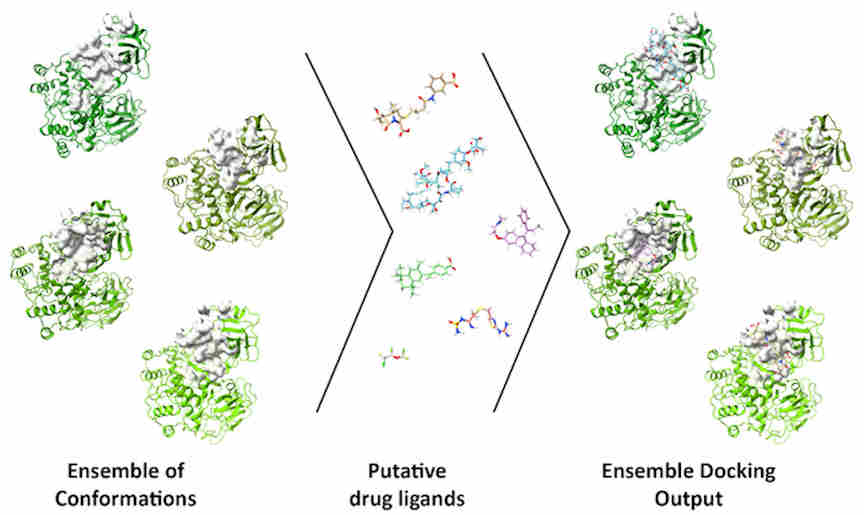
A research group has developed a web server to help researchers identify drugs to treat COVID-19. Their use of an ensemble of conformations allows researchers to account for protein flexibility in molecular docking studies.
Prepackaged protein binding sites and ensembles
The proteins currently available for docking through DINC-COVID are the main protease (Mpro), the papain-like protease (PLpro), and the RNA-dependent RNA polymerase (RdRp). For Mpro, protein structures are available for both the catalytic site and allosteric site.
"We chose [these four binding sites] because these can be targeted by different drugs," said Hall-Swan. "When you're trying to find a drug to inhibit a virus, you're going to look for the protein parts that are important for that virus to function and try to inhibit them."
The Mpro site in particular can substantially distort its shape in response to binding, allowing it to accommodate a diverse set of potential ligands, which is why the DINC-COVID team decided to make Mpro structures available for both the catalytic site and allosteric site.
"Unlike other servers, the proteins we are making available aren't static; they're not a single conformation," said Mauricio Rigo, PhD, a Rice University postdoctoral researcher and co-author of the paper. "We use states to reflect the dynamics of this protein in a physiological environment."
For each binding site, three ensembles are provided, including experimental data (i.e., structures from x-ray crystallography) or simulated data (i.e., structures from molecular dynamics with different force fields). The ensembles provide different scales of protein flexibility, from side-chain rearrangements to larger backbone motions.
The researchers decided to make precomputed ensembles available on the portal to facilitate the use of ensemble docking by a broad audience of users who are searching for new SARS-CoV-2 inhibitors but who are not necessarily computationally savvy.
"DINC-COVID allows users to run ensemble docking experiments without the additional burden of time-consuming simulations required for ensemble generation and docking preparation," they wrote.
Reception and future work
The response to DINC-COVID from the research community has been positive, according to the developers.
"We are very pleased with the response of the community to our work," Kavraki said. "DINC-COVID has already been used by about 500 researchers in 16 different countries, while our earlier web server DINC has been accessed by 11,000 users. We hope DINC-COVID will help shed light on the complex mechanisms of infection by SARS-CoV-2."
In future work on DINC-COVID, the researchers hope to expand the number of available ensembles, include the implementation of additional scoring functions during the sampling phase, and implement consensus ranking during the rescoring phase.
Do you have a unique perspective on your research related to bioinformatics? Contact the editor today to learn more.
---



Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


