



June 8, 2021 -- Scientists at the U.S. Department of Energy's Brookhaven National Laboratory have published the first detailed atomic-level model of the SARS-CoV-2 envelope protein in a study published June 8 in Nature Communications. The model shows how the virus interacts with lung proteins, helping to explain how SARS-CoV-2 causes extensive lung damage.
SARS-CoV-2 encodes a small envelope (E) protein that is critical for the assembly, release, and virulence of the virus. The protein is made up of 75 amino acids with two domains: an N-terminal transmembrane domain and a C-terminal domain.
The E protein also mediates host immune responses through activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome and PDZ-binding activity. The group of over 150 PDZ proteins is essential for regulating human immune responses.
Through an unknown mechanism, SARS-CoV-2 is able to hijack PDZ-domain-containing proteins in cell junctions to potentiate their virulence. It is thought that the E protein interacts with human cell junction protein PALS1, resulting in relocation of PALS1 from the cell junction and enabling viral assembly and maturation to occur.
"That interaction can be good for the virus, and very bad for humans -- especially elderly COVID-19 patients and those with preexisting medical conditions," said study lead author Qun Liu, PhD, a structural biologist at Brookhaven, in a statement.
When cell junctions are disrupted, leaking junctions promote viral spread and a flood of immune cells into the alveolar spaces in the lungs. This results in a massive inflammation event called cytokine storm that can lead to acute respiratory distress syndrome (ARDS) and severe COVID-19.
"In this scenario, most damage would occur in patients with more viruses and more E proteins being produced," Liu said.
For instance, more viruses make more E proteins and cause more cell-junction proteins to be pulled out, causing more damage, more transmission, and more viruses again. Additionally, any existing damage, such as lung-cell scarring, makes it harder for COVID-19 patients to recover from the damage.
"That's why we wanted to study this interaction -- to understand the atomic-level details of how E interacts with one of these human proteins to learn how to interrupt the interactions and reduce or block these severe effects," Liu continued.
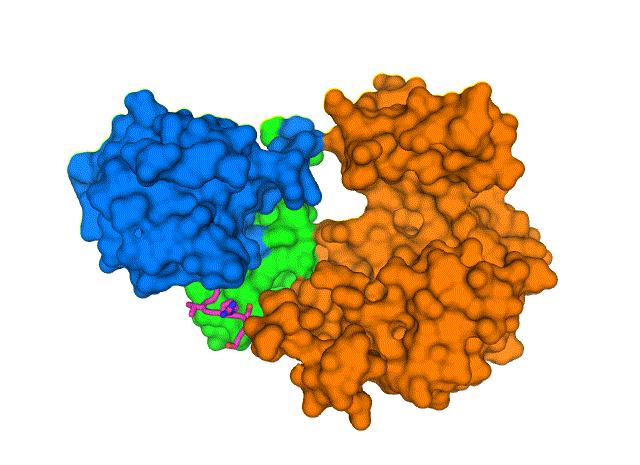
A new structure shows how the COVID-19 virus envelope protein (E, magenta sticks) interacts with a human cell-junction protein (PALS1; surfaces colored in blue, green, and orange). Understanding this complex structure, which was solved using a cryo-electron microscope at Brookhaven National Laboratory, could lead to the discovery of drugs that block the interaction and, potentially, the most severe effects of COVID-19. Image courtesy of Brookhaven National Laboratory.
To learn more about the structure of the E protein and its interactions with human lung cells, Brookhaven researchers used cryo-electron microscopy to determine the 3D map of protein-protein interactions and provide novel targets for potential inhibitors to block PALS1-E interactions and stop the spread of SARS-CoV-2 in the lungs.
"By obtaining atomic-level details of the protein interactions we can explain why the damage occurs, and search for inhibitors that can specifically block these interactions," Liu said. "If we can find inhibitors, then the virus won't cause nearly as much damage. That may give people with compromised health a much better chance for their immune systems to fight the virus successfully."
Advanced image-processing techniques allowed the team to solve the structure of the two proteins bound together. From the crystal structure, the researchers demonstrated that the E C-terminal PDZ-binding motif binds to a pocket formed by the PDZ and SRC homology 3 (SH3) domains. Moreover, SARS-CoV-2 encodes another protein, ORF3a that could be involved in the recruitment of PDZ-containing proteins that support its viral fitness and virulence.
"We can see how the chain of amino acids that makes up the PALS1 protein folds to form three structural components, or domains, and how the much smaller chain of amino acids that makes up the E protein fits in a hydrophobic pocket between two of those domains," Liu said.
They found that the PDZ and SH3 domains were severely rotated compared to normal physiological conditions, likely leading to the disengagement of the domains from cell junctions. This discovery suggests a mechanism for how the SARS-CoV-2 E protein is able to wrench PALS1 from its place at the cell's outer boundary.
SARS-CoV-2 drugs and evolution
"This structure provides the foundation for our computational science colleagues to run docking studies and molecular dynamics simulations to search for drugs or drug-like molecules that might block the interaction," said John Shanklin, PhD, a co-author and the chair of the biology department at Brookhaven. "And if they identify promising leads, we have the analytical capabilities to rapidly screen through such candidate drugs to identify ones that might be key to preventing severe consequences of COVID-19."
Furthermore, understanding the dynamics of this protein interaction will also help scientists track how viruses like SARS-CoV-2 evolve.
"This is one more reason it is so essential for us to identify and implement promising therapeutics," Liu explained. "In addition to preventing the most severe infections, drugs that effectively treat COVID-19 will keep us ahead of these mutations."
Do you have a unique perspective on your research related to virology or infectious disease? Contact the editor today to learn more.
---

Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


