



November 30, 2021 -- A group of researchers has developed a machine-learning framework that assigns the chemical shifts of organic crystals directly from their 2D structure -- accelerating an often time-consuming process. The study was published November 26 in Science Advances.
The breakthrough removes one of the main technical bottlenecks to more widespread application of solid-state nuclear magnetic resonance (NMR) spectroscopy, a technique used to determine the chemical structure, 3D structure, and dynamics of molecules and materials by measuring the frequencies emitted by the nuclei of atoms exposed to radio waves in a strong magnetic field.
"This method could significantly accelerate the study of materials by NMR by streamlining one of the essential first steps of these studies," said first author Manuel Cordova, a doctoral student at the Swiss Ecole Polytechnique Fédérale de Lausanne, in a statement.
The essential first step in question is called "chemical shift assignment," which involves assigning each peak in the NMR spectrum to a given atom in the molecule or material under investigation.
"Obtaining the assignment experimentally can be challenging and typically requires time-consuming multidimensional correlation experiments," the authors noted. "An alternative solution for determining the assignment involves statistical analysis of experimental chemical shift databases, but no such database exists for molecular solids."
Five years ago, the research team developed ShiftML, a machine-learning model that predicts chemical shifts in molecular solids. ShiftML is trained by comparing experimental chemical shifts with shifts computed using density functional theory, a computational quantum mechanical modeling method. After training, the system can generate accurate predictions on new structures without performing additional quantum calculations.
In the current paper, the researchers applied ShiftML to the Cambridge Structural Database (CSD), a database of more than 200,000 3D organic structures maintained by Cambridge University.
The researchers used ShiftML to predict shifts on more than 200,000 compounds extracted from the CSD, yielding statistical distributions of chemical shifts for each unique topological atomic environment. These environments were modeled as graphs, with vertices representing atoms and edges representing covalent connectivities. Atom pairs were identified as covalently bonded when the distance between the atoms in the pair were less than 1.1 times the sum of the covalent radii of the atoms involved.
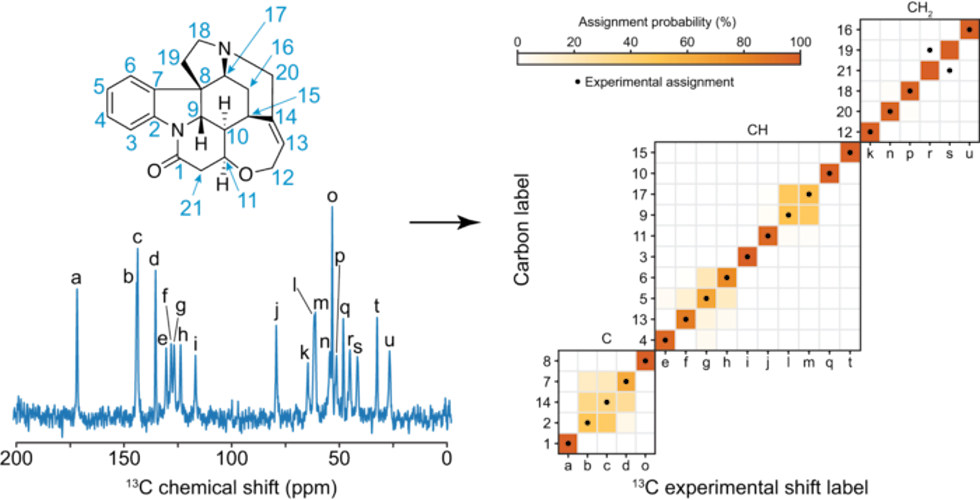
Probabilistic assignment of the 13C NMR spectrum of crystalline strychnine. Image courtesy of EPFL Manuel Cordova.
Because this simple graph representation based on covalent bonds does not encode any 3D structural features, it makes it possible to obtain the probabilistic assignment of the NMR spectra of organic crystals directly from their 2D chemical structures.
To make its assignment predictions, ShiftML performs Bayesian selection among the crystal structure prediction candidate structures that are compatible with the shifts measured by NMR and share the same graph as the corresponding atom in the molecule of interest.
The scientists tested their probabilistic framework by using ShiftML to predict the assignments on a set of 11 organic molecules for which the carbon chemical shift assignment had already, at least in part, been determined experimentally: theophylline, thymol, cocaine, strychnine, AZD5718, lisinopril, ritonavir, the K salt of penicillin G, β-piroxicam, decitabine, and simvastatin.
The results indicated that the assignments predicted by ShiftML generally aligned well with the experimentally determined ones. "The marginal individual assignment probabilities obtained directly from the 2D representation of the molecules were found to match the experimentally determined assignment in most cases," the authors reported in the paper. Furthermore, in cases where there were assignment ambiguities, the system narrowed down the candidates to a handful of possibilities for the NMR spectroscopist to further investigate.
Even in the case of the molecule strychnine, whose complex structure and crowded spectra makes shift assignment extremely difficult using traditional methods, the system correctly assigned 14 of the 21 unique carbon atoms with more than 75% confidence.
The researchers have published the Python source code and a user guide.
Do you have a unique perspective on your research related to machine learning or artificial intelligence? Contact the editor today to learn more.
---

Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


