



March 15, 2021 -- The SARS-CoV-2 virus has required little to no significant adaptation to humans since its jump from bats in late 2019, according to a new study published in PLOS Biology on March 12. The evolutionary analysis of coronaviruses in bats and humans reveals that the infectivity of SARS-CoV-2 is derived from adaptation in bats, not humans.
SARS-CoV-2 is the seventh known human-infecting member of the Coronaviridae family of RNA viruses. Genomic surveillance efforts have identified a subgenus of coronaviruses, called sarbecoviruses, that have been circulating in bat species in China.
Unlike other RNA viruses, sarbecoviruses can easily acquire adaptations, such as specific receptor-binding domain sequences and the inserted furin cleavage site, after switching to new host species for efficient replication and spreading. Specifically, SARS-CoV-2 exploits the generalist properties from acquired adaptations to increase angiotensin-converting enzyme 2 (ACE2) binding and facilitate infection of nonbat species, including humans.
Characterizing mutations can be useful to understanding the functional effect of genetic changes. Mutations under positive selection are more likely to reflect functional changes; however, frequency data associated with these mutations can be misleading, as growing populations tend to increase the average fitness of a virus. Viruses also undergo genetic drift, with deleterious mutations "surfing" expansion waves.
In the current study, an international team of researchers from the U.K., U.S., and Belgium investigated the evolutionary history of bat sarbecoviruses that shaped the emergence and rapid spread of SARS-CoV-2 by comparing positively selected mutations in the virus circulating in humans since the outbreak began to the signatures of historic selection acting on related bat viruses.
In total, the researchers analyzed 133,741 SARS-CoV-2 genomes sampled from December 2019 to October 2020 and compared them to 69 sarbecovirus genomes separated into phylogenetically congruent regions based on detected recombination patterns. The open-source analytical framework, Hypothesis Testing using Phylogenies (HyPhy), originally developed by study author Sergi Pond, PhD, professor of biology at Temple University, was instrumental in teasing out the signatures of evolution embedded in the virus genomes.
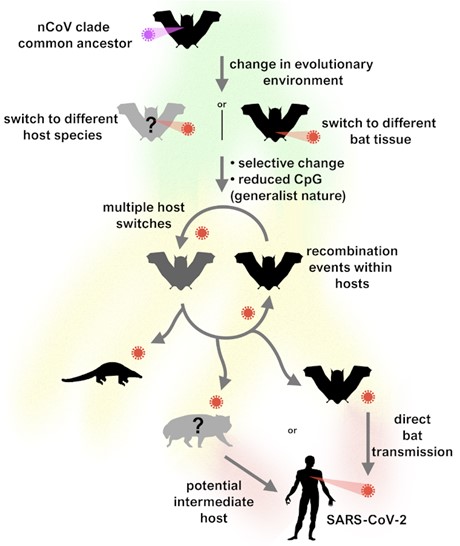
Schematic of the researchers' proposed evolutionary history of the new coronavirus clade and putative events leading to the emergence of SARS-CoV-2. Image courtesy of Oscar MacLean et al. Licensed under CC-BY.
"What's been so surprising is just how transmissible SARS-CoV-2 has been from the outset," Pond noted. "Usually, viruses that jump to a new host species take some time to acquire adaptations to be as capable as SARS-CoV-2 at spreading, and most never make it past that stage, resulting in dead-end spillovers or localized outbreaks."
Significant changes occurred to SARS-CoV-2, but before humans
Analysis of SARS-CoV-2 genomes across the timeline of the pandemic from December 2019 to October 2020 reveals a lack of diversifying selection taking place during the virus's circulation in humans. There is, however, evidence of ongoing positive selection associated with minor optimizations to the human population.
Still, the majority of adaptive changes that created the SARS-CoV-2 virus took place prior to its emergence in the human population.
"This does not mean no changes have occurred, mutations of no evolutionary significance accumulate and 'surf' along the millions of transmission events, like they do in all viruses," explained author Oscar MacLean, PhD, a bioinformatician at the University of Glasgow.
The data suggest that the SARS-CoV-2 progenitor is from a viral lineage with a relatively generalist nature. Specialization of viruses is driven by mutation rates and available opportunities for transmission, but at a price to generalism and the ability to replicate in multiple species.
"This stasis [neutral evolutionary changes] can be attributed to the highly susceptible nature of the human population to this new pathogen, with limited pressure from population immunity, and lack of containment, leading to exponential growth making almost every virus a winner," explained MacLean.
According to the authors, the ability of SARS-CoV-2 to readily transmit to other animals, such as pangolins and cats, is evidence of the virus's generalist property, which was evolved in bats. The lack of evidence of diversifying selection of immediately previous versions of SARS-CoV-2 prior to emergence in humans suggests that the adaptations occurred in bats that allow it to be a generalist capable of infecting humans and many other mammals and not in an intermediate host.
"Interestingly, one of the closer bat viruses, RmYN02, has an intriguing genome structure made up of both SARS-CoV-2-like and bat-virus-like segments," added Spyros Lytras, an author and doctoral candidate at the University of Glasgow Centre for Virus Research "Its genetic material carries both distinct composition signatures (associated with the action of host anti-viral immunity), supporting this change of evolutionary pace occurred in bats without the need for an intermediate animal species."
What about new variants since October 2020?
The authors noted that there are indications of increased selective pressure in recent variants of SARS-CoV-2 (such as the variants B.1.1.7 and B.1.351) that are associated with faster spread and a higher than usual number of nonsynonymous substitutions.
"[This] is because the immunological profile of the human population has changed," explained David Robertson, PhD, professor at the University of Glasgow Centre for Virus Research.
As the virus comes into contact with existing host immunity, variants that can dodge host responses are increasingly selected for. Coupled with the evasion of immunity in longer-term infections in chronic cases (e.g., in immunocompromised patients), these new selective pressures are increasing the number of important virus mutants.
Due to the high diversity and generalist nature of sarbecoviruses, a future spillover, potentially coupled with a recombination event with SARS-CoV-2, is possible, according to the researchers. The potential emergence of a "SARS-CoV-3" could evade either natural or vaccine-acquired immunity. Therefore, the authors advocate for increased surveillance for sarbecoviruses at the human–animal interface and monitor carefully for future SARS-CoV emergence in the human population.
Do you have a unique perspective on your research related to bioinformatics or genomics? Contact the editor today to learn more.
---


Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


