



August 1, 2022 -- Newborn neurons struggle to reach their proper places in advanced human brain tissue models of Rett syndrome, new research finds. This shows how developmental deficits may emerge in people with Rett syndrome.
The disorder is characterized by severe intellectual disability and impaired social behavior due to mutations in the MECP2 gene. Scientists at the Picower Institute for Learning and Memory at Massachusetts Institute of Technology learned more about the mutation by growing 3D cell cultures, also known as "minibrains" using cells from people with MECP2 mutations. They compared these cerebral organoids with otherwise identical cultures without the mutations (eLife, July 29, 2022).
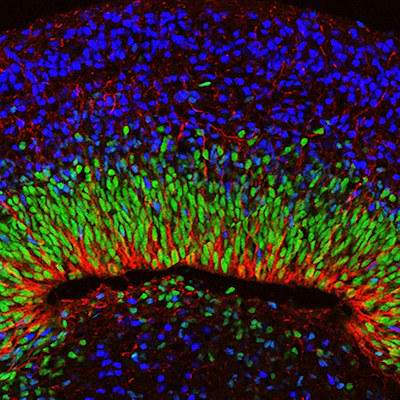
Using third harmonic generation (THG) three-photon microscopy, the team was able to image the organoids without damaging tissues or adding any chemicals to label cells. The microscopy technique uses a laser but applies less than 5 milliwatts of power, which is equivalent to a cat toy laser pointer.
The researchers validated their results by comparing THG images to images made via more traditional chemical labeling methods. They found the nascent neurons in the minibrains modeling Rett syndrome moved slowly from the rim around ventricles in the minibrains to the outer edge, analogous to the brain's cortex. The cells also "meandered" when compared with the faster motion in straighter lines shown by the same cell types in organoids without MECP2 mutation.
In Rett syndrome organoids, the ventricles were larger, more numerous, and thinner. Using THG, they also found Rett syndrome neurons achieved only about two-thirds the speed of non-mutated neurons and were significantly more "wiggly."
Up next, the team plans to investigate how MECP2 influences genes and molecules that affect neuronal migration.
Copyright © 2022 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


