



July 13, 2021 -- Two new studies point to the efficient use of gene therapy to treat deadly childhood genetic diseases. The first study used direct gene therapy infusion to treat a rare neurodevelopmental disorder, while the second study used prenatal base editing in mouse models as proof of concept for the treatment of genetic diseases. These vastly different approaches show the promise of gene therapy for a variety of devastating diseases.
Gene therapy No. 1: Direct DDC gene therapy via viral vector
Aromatic L-amino acid decarboxylase (AADC) deficiency is a rare neurodevelopmental disorder characterized by the deficient synthesis of dopamine to serotonin. The deficiency impacts only 135 children worldwide and is caused by mutations in the DOPA decarboxylase (DDC) gene, which leads to a dysfunctional AADC enzyme. It causes physical and developmental problems, including intellectual disability in at least 70% of patients.
An early-stage clinical trial led by researchers at Ohio State University Wexner Medical Center and Ohio State University College of Medicine applied direct gene therapy (gene replacement with functional genes) to seven children ages 4-9 with AADC deficiency. The results of the study were published in Nature Communications on July 12.
The corrected gene (human AADC complementary DNA; hAADC) was packaged into an adeno-associated virus, serotype 2 (AAV2) vector. A direct infusion of AAV2-hAADC was administered into two regions of the midbrain. Dopaminergic neurons in these regions send branched projections into other regions of the brain. Therefore, the axonal transport of viral vectors was able to disseminate the therapeutic gene along brain circuitry affected by the gene mutation.
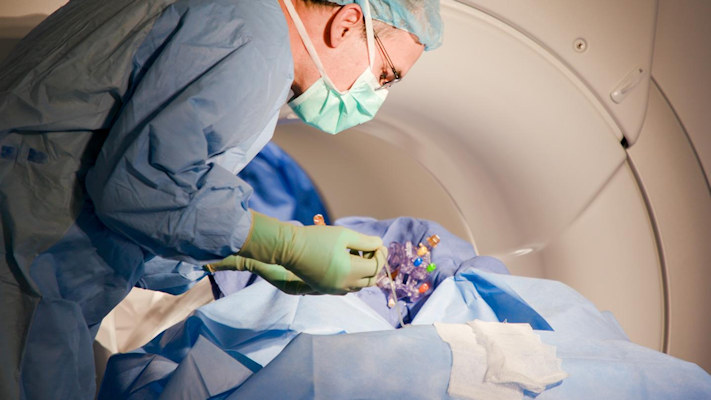
The direct gene therapy was infused into the specific regions of the brain of the subjects via an infusion catheter. Then, the researchers used real-time magnetic resonance imaging (MRI) to confirm vector delivery.
"Really, what we're doing is introducing a different code to the cell," said Dr. James Elder, director of neurosurgical oncology at Ohio State Wexner Medical Center's Neurological Institute, in a statement. "And we're watching the whole thing happen live. So, we continuously repeat the MRI and we can see the infusion blossom within the desired nucleus."
 .
.
Dr. James Elder performs a gene therapy brain infusion at The Ohio State University Wexner Medical Center. Image courtesy of The Ohio State University Wexner Medical Center.
All seven trial subjects exhibited measurable improvements in clinical function and dopamine production, suggesting that restoration of AADC activity in physiological regions (midbrain) can reverse the pathologic phenotype observed in primary AADC deficiency.
"Remarkably, these episodes are the first symptom to disappear after gene therapy surgery, and they never return," said co-author Dr. Krystof Bankiewicz, PhD, professor of neurological surgery at Ohio State College of Medicine. "In the months that follow, many patients experience life-changing improvements. Not only do they begin laughing and have improved mood, but many are able to begin speaking and even walking. They are making up for the time they lost during their abnormal development."
The team believes that the methodology and results from the trial provide a framework for the treatment of other neurodegenerative diseases such as Parkinson's disease and Alzheimer's disease.
Gene therapy No. 2: Prenatal base editing
Hurler syndrome, also known as mucopolysaccharidosis type I (MPS-IH), is a lysosomal storage disease that affects around one in 100,000 infants globally. The disease is caused by a single DNA base mutation, where adenine replaces guanine. The disease presents in children under 6 months of age with swelling of the liver and spleen, abdominal wall hernias, musculoskeletal abnormalities, retinal and neurocognitive degeneration, and cardiac disease. If untreated, patients can die of cardiorespiratory complications by 5 to 10 years of age.
Current postnatal treatments for children include lifelong immunogenic enzyme replacement therapy and hematopoietic stem cell transplantation. Postnatal systemic gene therapy strategies are also currently under investigation but still have inefficiencies and safety issues. By contrast, CRISPR base editing may offer a potentially safe and more efficient solution.
Although MPS-IH typically presents symptoms by 6 months of age, it can be prenatally diagnosed. The timing of the onset of the disease suggests that in utero base editing may offer the potential to treat MPS-IH prior to birth.
With an entirely different approach, researchers from the Children's Hospital of Philadelphia (CHOP) used a DNA base editing technique in a prenatal mouse model to correct Hurler syndrome. The adenine base editor targeting the guanine to adenine mutation in Hurler mice was delivered with an AAV9 in utero. The results of the study were published in Nature Communications on July 13.
"This study shows that prenatal base editing for Hurler syndrome is feasible in a preclinical mouse model," said senior author Dr. William Peranteau, attending surgeon at CHOP and chair of the university's fetal and pediatric surgery department. "In addition to showing the benefit of treating the disease before birth, we also showed some correction of the disease with base editing after birth, highlighting the promise of both pre- and postnatal base editing for Hurler syndrome."
The base editing strategy was shown to correct the disease-causing mutation in multiple disease-affected organs in the mouse model, with a high-level correction in the liver and heart. Prenatal base editing was associated with the amelioration of the cardiac and musculoskeletal phenotypes at 6 months of age. Control mice experienced cardiac decline between 4 to 6 months, sometimes leading to death.
An important advantage of prenatal base editing is the potential immunologic benefit of a less immunogenic AAV delivery mechanism. This offers the possibility of delivering an additional postnatal booster therapy.
The researchers found that postnatally treated mice also demonstrated efficient on-target editing in the heart and liver. This points to the potential benefits of base editing as a postnatal treatment; however, the authors noted that the maximum effect may be realized with earlier therapy.
The authors also noted several areas for improvement before the results of the study could be clinically translatable. For instance, AAV toxicity must be thoroughly assessed prior to clinical evaluation and strategies to enhance transduction may improve editing efficiency.
"Although the safety of these approaches for mothers and fetuses still needs to be rigorously characterized prior to clinical translation, this proof-of-concept study offers hope for genetic diseases with limited postnatal treatments," Peranteau said.
Do you have a unique perspective on your research related to gene therapy? Contact the editor today to learn more.
---



Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


