



March 25, 2020 -- Drug resistance is a common challenge when designing therapeutics for cancers or diseases originating from bacteria or viruses. Researchers explored how evolution impacted the mutation of these pathogens, and their findings could lead to the development of evolutionarily designed drugs. The results were published March 24 in Cell Reports.
Rational drug design -- or the development of medication based on the study of the structures and functions of target molecules -- has been relatively successful. However, once mutations begin to change cells, new treatments are required.
This drug resistance is a challenge when treating diseases caused by bacteria, viruses, or cancers. In order to develop new drugs, developers must target appropriate mutations to kill pathogens or cancer cells.
"We need to not just understand the biophysics," said Justin Pritchard, PhD, assistant professor of biomedical engineering at Penn State University. "We also need to understand the evolutionary dynamics."
Penn State University researchers wanted to understand what drives mutations in the real world and, if they could predict which mutations are most likely to occur, create an easier path to making effective pharmaceuticals.
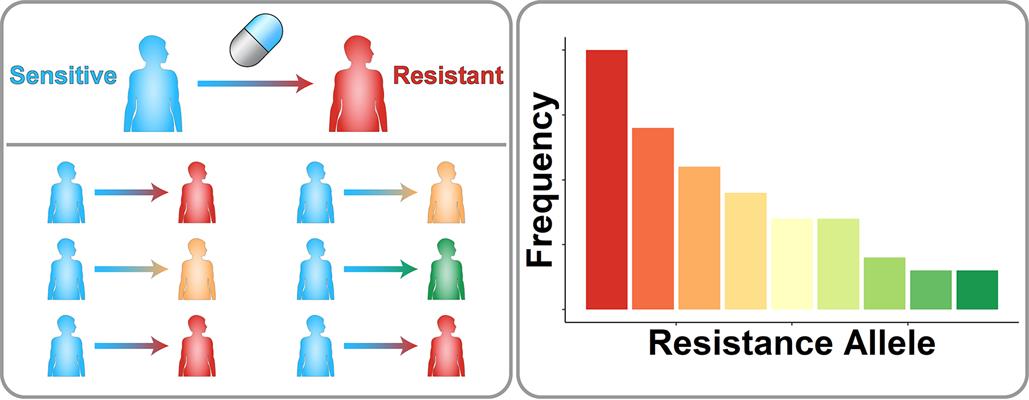
(Left) A schematic of drug resistance across a population of patients. Patients with initially sensitive disease (blue) are treated with a drug. Different genetic mutations cause different resistance mutations (red, yellow, and green). (Right) Tallying up the number of patients associated with each resistance allele shows that some alleles are more common in the clinical population than others. Image courtesy of Scott Leighow, Penn State.
To study this, the researchers focused on cancers because cancer cells are a simpler model, are not contagious, and do not have additional potential sources of mutations. They chose to look at leukemia data because the database was the largest and most complete.
BCR-ABL is a mutation formed by the combination of two genes, BCR and ABL, that commonly occurs in patients with leukemia. The oncogene of BCR-ABL, E255, forms a salt bridge interaction with K247 to stabilize a phosphate-binding loop that blocks an ATP competitive inhibitor in the active site of the molecule. E255 can mutate to become either E255V or E255K to promote resistance to imatinib, a BCR-ABL tyrosine kinase inhibitor to treat patients with chronic myelogenous leukemia.
The researchers found that E255K was more prevalent than E255V. Both mutations provided resistance to imatinib, but E255K provided significantly less resistance than E255V. Therefore, they sought to understand how a mutation that is worse at conferring drug resistance might become more prevalent in humans.
"We shouldn't always focus on the strongest resistance mutation because there are other evolutionary forces that dictate what happens in the real world," said Pritchard. "Sometimes drug resistance relies on biased random events."
This is partially explained by the likelihood of specific amino acid changes of E255V (AT) versus E255K (GA). Simulations revealed that G to A (resulting in K variant) mutations were more likely than A to T (V variant) mutations. Moreover, the substitution likelihood of a given amino acid mutation (defined as the nucleotide substitution bias and the individual codon path) is correlated with the clinical prevalence of E255V versus E255K.
When analyzing clinical data, researchers found that the 19 most abundant amino acid mutations accounted for around 95% of the resistance mutations that occurred. A statistical model developed by the researchers suggested that the degree of resistance and the substitution likelihood of a particular variant were both predictors of clinical abundance; however, substitution likelihood was more significant.
The researchers conceived a hypothetical BCR-ABL inhibitor, which they called maxitinib, that they designed via structure-based drug design to target the five most likely mutations. They simulated in silico application of maxitinib for leukemia patients.
The model predicted a 67% reduction in resistance with maxitinib relative to imatinib. This suggests that second-generation inhibitors with an evolution-guided approach could minimize drug resistance across a population.
The team found similar results in simulations of breast, prostate, and stomach cancers, although the datasets were not as large.
"The data are not quite as strong in the prostate and breast cancer setting," said Pritchard. "In non-small cell lung cancer, we didn't see this effect at all."
In many cases, evolutionary bias creates an abundance of mutations that are not the most resistant strains. In oncology, a spectrum exists with leukemias on one end and non-small lung cancer on the other end.
"Our analysis establishes a principle for rational drug design: When evolution favors the most probable mutant, so should drug design," according to the researchers.
Do you have a unique perspective on your research related to drug discovery and development? Contact the editor today to learn more.
---
Copyright © 2020 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


