



April 27, 2021 -- Researchers have developed a novel chemical tool for elucidating protein interaction networks within cells. The technique, details of which were published in Molecular Cell on April 23, allows for the visualization of protein-protein interactions.
Posttranslational modifications of proteins signal downstream events that play key roles in regulation of many cellular processes. Modified proteins alter the physical and chemical proteins of proteins that change their subcellular localization and biological function.
Posttranslational modifications can also serve as docking sites for downstream effector proteins, called "readers," that regulate cellular processes. Therefore, dysregulation of these protein-protein interactions can lead to diseases such as cancer or neurodegenerative diseases.
Because many posttranslational modifications are highly dynamic, and mediate weak or transient protein-protein interactions, traditional methods for studying protein interactions, such as affinity purification or hybrid screening, are limited.
Photo-crosslinking-based methods that incorporate affinity tags (biotin) or biorthogonal handles (alkyne) onto probes have been developed to identify posttranslational modifications such as methylation and phosphorylation, among others. These methods provide the opportunity to map protein-protein interactions in relation to each other. However, mapping binding regions of posttranslational modifications with photoaffinity probes remains a challenge.
To dissect complex protein networks, researchers from the University of Hong Kong designed a tri-functional amino acid, called alkynyl diazirino disulfanyl cysteine (ADdis-Cys). The team wanted to elucidate the "who" and "how" of protein binding of posttranslational modifications. In this case, the "who" are the interacting proteins, and the "how" is the specific binding sites that facilitate the protein interactions.
This photo-crossslinkable molecule can convert noncovalent (transient) interactions into irreversible covalent linkages upon ultraviolet (UV) irradiation. Affinity tags can be added to the alkyne to facilitate the isolation of crosslinked (trapped) proteins. The disulfide moiety allows for the release of crosslinked proteins during coupled mass spectrometry analysis.
Cumulatively, the tri-functional amino acid in combination with mass spectrometry (ADdis-Cys-MS) can be used to identify posttranslational modification readers from complex proteomes and to map binding sites of protein-protein interactions.
Comprehensive identification of protein interactions
The researchers focused on using photoaffinity probes to recognize histone posttranslational modifications and identification of histone posttranslational modification binding sites using ADdis-Cys-MS. The first probe they developed was designed to trap known protein readers of trimethylation at histone H3 lysine 4 (H3K4me3).
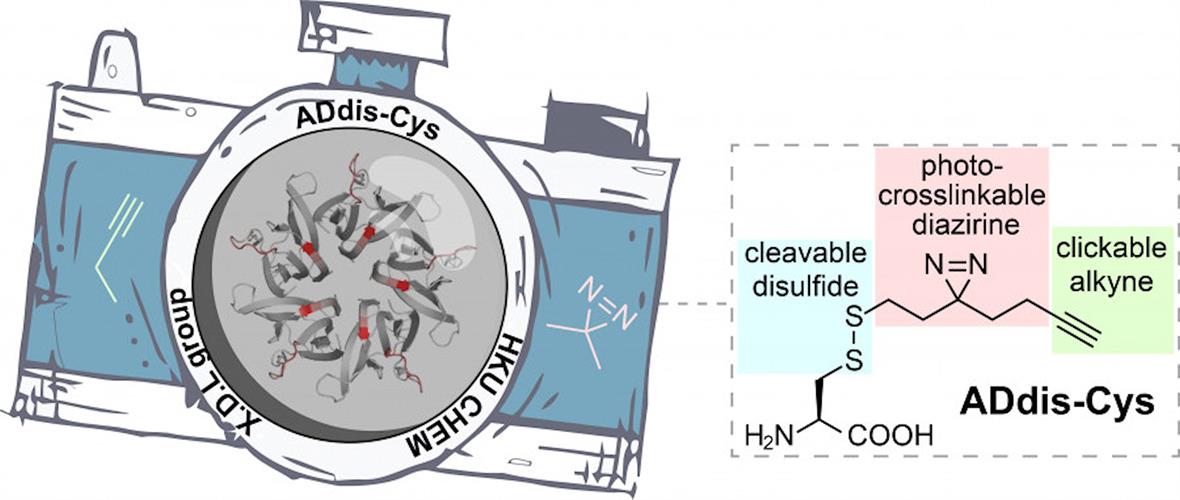
The ADdis-Cys "camera" can simultaneously identify a protein's interacting partners and pinpoint their binding regions. Image courtesy of the University of Hong Kong.
The protein readers were incubated with the probe and irradiated using UV light. The labeled proteins were resolved using gel electrophoresis and detected using in-gel fluorescence scanning. The results confirmed that the probe could selectively and efficiently bind to specific protein readers.
Next, to begin to understand how probes could be used to detect binding sites, the team used ADdis-Cys to tag the SPIN1 protein, which binds to H3K4me3 via a tandem Tudor domain. The probed protein was analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for retention of the alkyne tag. The researchers mapped MS results onto a reported crystal structure of SPIN1-H3K4me3 peptide complex and found that all the crosslinked residues were located in SPIN1's H3 peptide-binding pocket, near when ADdis-Cys was installed.
For many posttranslational modifications, structural information is missing. So, the researchers used ADdis-Cys-MS to reveal posttranslational medication-binding sites of readers that lack structural information. They used a zinc finger myeloid, Nervy, and DEAF-1 (ZMYND)-type containing 8 (ZMYND8) that recognizes a dual histone modification H3K4me0/1-H3K14ac. Posttranslational modifications of ZMYND8 rely on the triple reader module plant homeodomain-Bromo-Pro-Trp-Trp-Pro (PHD-Bromo-PWWP).
Although the crystal structure of PHD-Bromo-PWWP has been solved, there is no structure for the binding complex with 3K4me0/1-H3K14ac. They labeled ZMYND8 with peptide probes and mapped the identified residues to the crystal structure of the ZMYND8 PHD-Bromo-PWWP module. They found that one probe was specifically bound in the PHD domain, whereas the other probe showed a preference for crosslinking to the bromodomain (BrD).
In their analysis, the researchers also identified complement component 1 Q subcomponent-binding protein (C1QBP) as a novel histone chaperone using the disordered domains (lacking fixed 3D structures).
Altogether, ADdis-Cys-based probes are able to label known binding proteins, but also help identify protein-posttranslational modification interacting surfaces that correlate to resolved protein crystal structures. The tri-functional amino acid was also useful in determining the molecular basis of posttranslational modification interactions that lack crystallographic information.
This tool could lead to the development of chemical modulators that regulate protein interactions for the treatment of deadly human diseases. ADdis-Cys may also have wide-reaching applications in many areas of biomedical and biopharmaceutical research.
Do you have a unique perspective on your research related to proteomics? Contact the editor today to learn more.
---

Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


