


October 12, 2021 -- Researchers have developed an algorithm that can take either single-cell mass cytometry data or RNA sequencing data, align it, and extract a common denominator for comparing cancer data among patients. The algorithm, called tuMap, was detailed in a study published on October 7 in Cell Systems.
The tuMap algorithm takes single-cell trajectories and aligns them to a reference point, enabling cancer samples to be compared at high resolution to identify prognostic markers and predict clinical indices. The algorithm was developed by researchers at the Technion Israel Institute of Technology, the Rambam Health Care Campus, the Shaare Zedek Medical Center, and the University of Texas.
The problem of interpatient variability
Single-cell technologies are used by cancer researchers to measure and understand variability among tissues. Technologies such as single-cell RNA sequencing and mass cytometry provide high-resolution characterization of the abnormal developmental processes observed in cancer.
While these tools provide researchers with vast amounts of data characterizing the cells, the data also has high variability due to the high variability in tumors of different patients, which has traditionally prevented systematic comparison among the data. In the absence of a common denominator, there is no way to compare datasets to estimate mortality, the severity of illness, or other prognostic markers.
Building a comparative framework
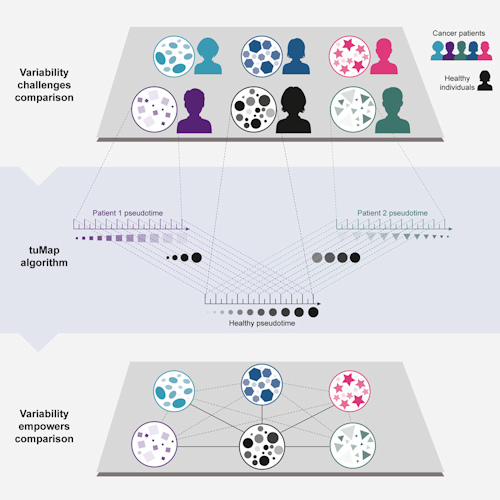
The work was based on three datasets: one from healthy individuals and two from leukemia patients. The healthy dataset consisted of single-cell mass cytometry data sampled from 10,000 cells per individual from a set of bone marrow samples from five healthy individuals.
The first leukemia bone marrow dataset comprised single-cell mass cytometry data from acute lymphoblastic leukemia patients. The second consisted of single-cell mass cytometry data, as well as single-cell RNA sequencing data, from acute myeloid leukemia (AML) patients.
Graphical concept of tuMap algorithm. Image courtesy of Alpert et al. Licensed under CC-BY-NC-ND.
The researchers then aligned the cancer samples to the healthy developmental axis on a single-cell basis using the psupertime algorithm, a semi-supervised method that performs cellular classification into cell populations followed by optimization of their ordering. After experimenting with other algorithms, the team found that psupertime, which is available as a package on the R data processing platform, was best able to extract the healthy developmental axis from the cancer samples.
Once the alignment was completed, the next step was pseudotime analysis, in which the high-dimensional molecular data from the cross-sectional cohort of individuals was mapped onto a series of one-dimensional quantities called pseudotimes. Pseudotime is a latent (unobserved) dimension that measures the cells' developmental progress in a single, consistent manner.
The result of the analysis was the tuMap pseudotime axis, a level of representation that is similarly scaled across all samples. The pseudotime axis allows systematic and meaningful comparisons of high-dimensional, single-cell data across cancer patients within a single cancer patient over time (for example, the timeline from diagnosis to post-treatment) and against a healthy counterpart.
Clinical applications
To demonstrate the clinical value of the tuMap pseudotime axis, the researchers used it to create survival models to predict relapse occurrence in a longitudinal clinical dataset. The tuMap approach yielded better incremental area under curve and c-statistic values than a baseline method that used averaging by coarse-grained cellular grouping into cell populations, suggesting that the tuMap approach can uncover clinically meaningful information that may be masked by stratification into cell populations.
In addition, using the single-cell RNA sequencing dataset from the AML patients, the researchers used the tuMap pseudotime axis to identify very early leukemic stem cells, thus showing the potential of the approach to cancer prediction.
"A comparative framework for these samples is an essential tool for tumor characterization of patients to forward a deeper understanding of molecular dynamic mechanisms and fine-tuned clinical metrics that are the building blocks of a cancer precision medicine era," the authors concluded.
Do you have a unique perspective on your research related to single-cell technologies? Contact the editor today to learn more.
---
Copyright © 2021 scienceboard.net