


August 31, 2021 -- A new alternative splicing system called Xon can modulate levels of protein expression in gene therapy, much like a dimmer switch. The invention addresses a major shortcoming of most gene therapies, which is the ability to regulate gene expression levels in diseases like spinal muscular atrophy (SMA).
SMA is a rare neuromuscular disorder. It is caused by mutations in the SMN1 gene, which encodes a protein called survival of motor neuron (SMN). As the name suggests, SMN is important for the survival of neurons, and the lack of SMN leads to the loss of motor neurons and progressive muscle wasting. SMA is an autosomal recessive inherited disorder, meaning both copies of the inherited gene (one from each parent) must be defective. Most humans have at least one copy (usually two to four copies) of a second gene called SMN2, which also encodes SMN. However, that gene is not efficiently translated into protein due to a process called alternative splicing.
RNA splicing plays an important role in gene regulation. Genes are typically interspersed with several noncoding regions called introns that interrupt the exons that make up the final mRNA transcript. The initial mRNA transcript, which is called a pre-mRNA molecule, includes both introns and exons, and the process of RNA splicing removes the introns to leave a mature mRNA transcript (composed only of exons) to be translated into protein.
A single gene can give rise to multiple proteins though alternative splicing, where different introns are removed from the same pre-mRNA to create differently spliced mature mRNAs.
Although SMN2 is almost identical to SMN1, a variation in a single nucleotide between the two genes results in very different mRNA splicing patterns. Only about 10% to 20% of SMN2 gene transcripts are spliced into mature mRNAs coding for full-length SMN, while the remainder result in the deletion of exon 7 to encode a truncated version of the protein called SMNΔ7. The truncated form is degraded within the cell almost immediately, but the small percentage of SMN2 mRNAs which are successfully translated into full-length SMN protein can lessen the severity of SMA (caused by the absence of a functional SMN1 gene).
In general, the more copies of the SMN2 gene an individual possesses, the less severe their SMA symptoms will be.
LMI070 (also known as branaplam) belongs to a class of drugs called splice modulators, which can alter the splicing pattern of mRNA transcripts. The orally bioavailable, small-molecule drug modifies the splicing pattern of SMN2 mRNA to include exon 7, thereby increasing the expression of the full-length SMN protein. Novartis began phase I/II clinical trials for LMI070 in 2015 for the treatment of severe SMA in children with two copies of the SMN2 gene.
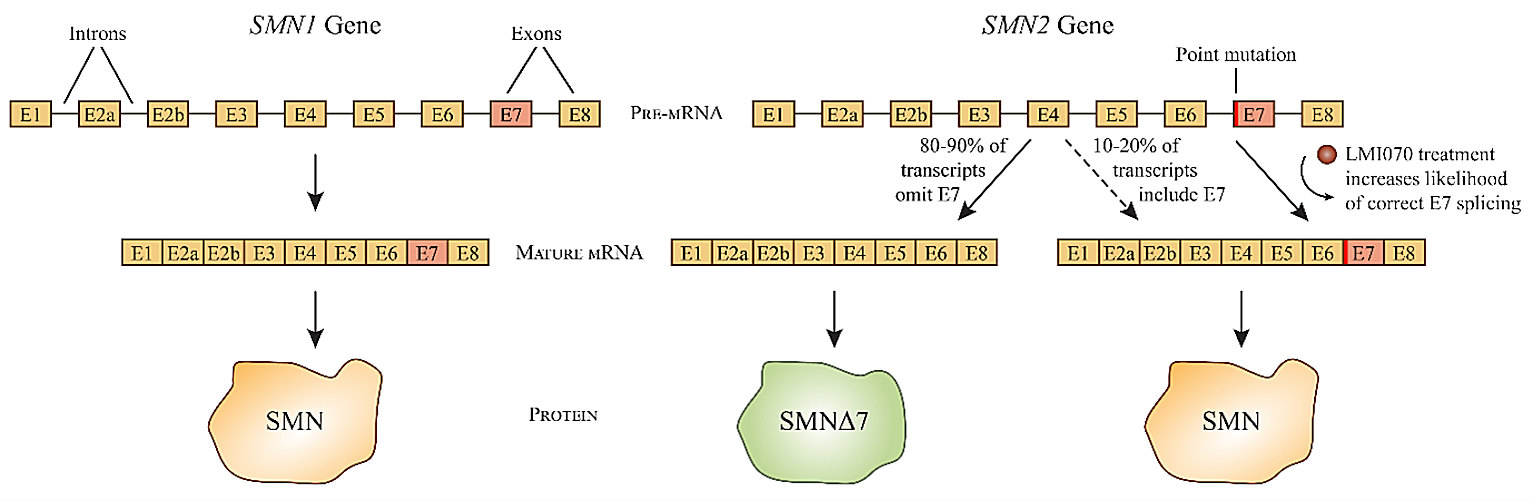
Schematic showing splicing patterns of the SMN1 and SMN2 genes. While the SMN1 gene's pre-RNA is always spliced to include exon 7, a point mutation in the SMN2 gene results in the deletion of exon 7 from the majority of mature mRNA transcripts. As a result, mRNAs transcribed from the SMN1 gene are translated into full-length SMN protein, while most of those transcribed from the SMN2 gene are translated into a truncated protein, SMNΔ7. Treatment with LMI070 restores correct splicing of exon 7 in SMN2 gene mRNA transcripts. Image courtesy of Science and Medicine Group.
Although Novartis has since abandoned development of LMI070 in favor of newer therapeutics for the treatment of SMA, clinical trial data from the SMA study indicated that the drug also decreased expression of huntingtin protein, an important therapeutic target for Huntington's disease. Novartis began clinical trials for LMI070 in the treatment of Huntington's disease this year.
Modifying splice modulators to develop Xon
Now, Children's Hospital of Philadelphia (CHOP) scientists, in collaboration with Novartis researchers, decided to see if they could adapt LMI070's RNA splicing capabilities to regulate the expression of other genes.
Initial in vitro studies were conducted by adapting SMN2's exon 6/7/8 region into an expression cassette to control the expression of any gene cloned into it. After success with that construct, they next looked for other exons that might be more sensitive to LMI070 by treating cultured cells with the drug and using high-throughput RNA sequencing to discover which genes were most actively expressed by the treatment.
Several likely exon candidates were tested in vitro using expression cassettes designed to include the translation start codon within the alternatively spliced exon so that protein translation could not begin unless the drug induced successful splicing of that exon into the mature mRNA.
In the end, exon sequences from a gene called SF3B3 (encoding subunit 3 of the splicing factor 3b protein complex) demonstrated over a hundredfold induction over baseline (more than fivefold greater than SMN2's switch) and were chosen as the basis for their tunable switch system, Xon.
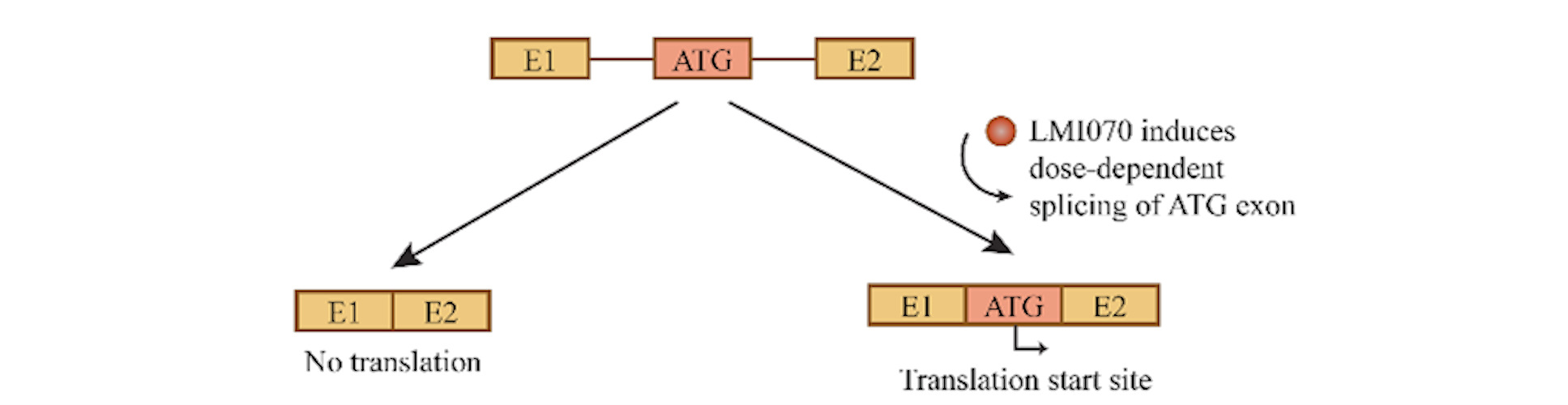
The Xon system, which uses exons adapted from the SF3B3 gene. Since the translation start codon (ATG) is included in the alternatively spliced exon, translation cannot begin unless splicing of that exon into the mature mRNA is induced by LMI070.
The Xon system was next tested in vivo in mice, first using a reporter gene (firefly luciferase) to show that gene expression could not only be activated by LMI070 but also controlled in a dose-dependent manner -- the higher the dose of LMI070, the more luciferase was expressed.
From there, the researchers tested the system in actual gene therapy experiments. Mice were treated with a recombinant adeno-associated vector (AAV) containing an Xon expression cassette encoding erythropoietin, a kidney protein important for the production of red blood cells. The mice were then administered LMI070 in varying doses, and scientists were able observe dose-responsive effects on both expression and therapeutic effect. When LMI070 treatment was suspended, the levels of erythropoietin would eventually return to baseline, but expression could be later reactivated by subsequent dosing.
Improving Xon with CRISPR
The Xon system is particularly advantageous in CRISPR-Cas9 gene editing therapies, where the expression of the DNA editor is only required for a brief time. After the therapeutic edits have been made, continued expression is not desirable, as it can lead to immunological reactions and off-target edits. The CHOP team was able to demonstrate the feasibility of switchable gene editing in mice using the Xon system to control SaCas9, a smaller version of Cas9 derived from Staphylococcus aureus.
In another mouse experiment, researchers were able to use the Xon system to control the expression of progranulin in the brain. Progranulin is a growth factor that regulates neuronal outgrowth and survival, but treating diseases caused by its deficiency can be tricky, as the protein quickly becomes toxic if the therapeutic dosage is exceeded. The Xon system was able to precisely control the dosage delivered.
"This study shows that by using a splicing modulator in combination with gene therapy tools, the dose of protein expressed from gene therapy vectors can be controlled for maximum therapeutic benefit," said lead author Beverly Davidson, PhD, professor at CHOP, in a statement.
Novartis has already licensed the technology and plans to continue collaborating with CHOP to develop Xon and other small-molecule splicing modulators that can be used to finely adjust gene expression levels in a variety of clinical applications.
Blake Middleton is the editor of Cell and Gene Therapy Business Outlook, part of Science and Medicine Group.
Disclosure: Cell and Gene Therapy Business Outlook is a sister publication of ScienceBoard.net.
Do you have a unique perspective on your research related to gene therapy? Contact the editor today to learn more.
---
Copyright © 2021 scienceboard.net