


July 29, 2021 -- Using machine learning, researchers have developed a method that evaluates the contributions of all possible tumor mutations to the development and progression of cancer. The results of the study were published in Nature on July 28.
Most mutations observed in cancer genes across many tumor types are of unknown significance to tumor growth. The increased ability to sequence tumors has resulted in the knowledge that more than 500 genes are under positive selection in various types of tumors. However, researchers still have difficulty distinguishing driver mutations from passenger mutations in cancer genes. Therefore, determining the relevance of the 90% of cancer gene variants of unknown significance could have clinical relevance in patient tumor samples.
Researchers from the Institute for Research in Biomedicine (IRB Barcelona) and the Catalan Institution for Research and Advanced Studies (ICREA) collected somatic mutations of approximately 28,000 tumors across 66 cancer types and identified 568 mutational cancer genes. By restricting the dataset to cancer genes in a tissue with mutations associated with oncogenic mechanisms, the researchers identified 282 gene-tissue combinations. Using these known oncogenic mutations, the team trained machine-learning models (called BoostDM) to establish a discovery index as a measure of the representativeness of a mutation within a cancer gene in a tumor type for its potential driver status.
"BoostDM goes further: It simulates each possible mutation within each gene for a specific type of cancer and indicates which ones are key in the cancer process," said Núria López-Bigas, PhD, head of the biomedical genomics lab at IRB Barcelona, in a statement. "This information helps us to understand how a tumor is caused at the molecular level, and it can facilitate medical decisions regarding the most appropriate therapy for a patient."
Traditionally, saturation mutagenesis (a method whereby a single codon is substituted with all possible amino acids at a specific position of a gene) is used to experimentally classify driver mutations. BoostDM showed the ability to computationally classify experimentally validated rate mutations in oncogenes. Further, all BoostDM models outperformed the results of saturation mutagenesis assays and seven other computational methods designed to identify driver mutations or assess the functional impact of mutations.
From evolutionary biology
Tumors follow Darwinian evolution, where genes under positive selection (those that promote tumor growth) are found in higher numbers, compared to mutations that occur by random chance.
"We started from the premise that we only get to observe some mutations because the tumor cells with this mutation guide the development of the tumor, and we questioned what distinguishes these mutations from other possible mutations, " explained co-author Ferran Muiños, PhD, who is a postdoctoral researcher at IRB Barcelona. "Doing this analysis manually would be excessively laborious, but there are computational strategies that allow it to be organized systematically and efficiently."
The computational method learns what attributes are distinctive of the mutations that favor the development of cancer and leverages them for therapeutic development.
Gene-tumor specific models
Determining cancer driver mutations with high-throughput experimental mutagenesis is a massive undertaking. Bioinformatics methods have been developed to speed the process but remain difficult given the different molecular mechanisms of tumorigenesis for each cancer gene and tissue.
BoostDM, however, was able to identify how the same gene mutations affect different tumor types. For instance, the approach generated a model that identified all the possible mutations in the epidermal growth factor receptor (EGFR) gene that triggers tumor development in some lung cancers, another model for the same gene in cases of glioblastoma, and so on.
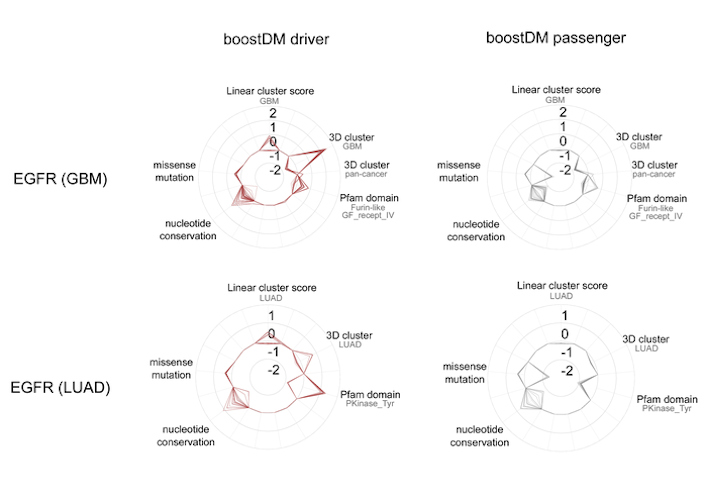
This diagram displays the contribution of mutational features to the classification of all EGFR driver and passenger mutations in lung adenocarcinomas and glioblastomas. Image courtesy of IRB Barcelona.
Ultimately, the model will be able to undercover many potential driver mutations that have not been previously identified in tumors. The technology could also be instrumental for classification of mutations observed in patient tumor samples for precision medicine applications, according to the authors.
The BoostDM tool has been integrated into the IntOGen platform, developed by the same group and designed to be used by the scientific and medical community in research projects. It has also been integrated into the Cancer Genome Interpreter, also developed by this group and which is more focused on clinical decision-making by medical oncologists.
Do you have a unique perspective on your research related to artificial intelligence or cancer research? Contact the editor today to learn more.
---
Copyright © 2021 scienceboard.net