


June 3, 2020 -- A new technique has shown that conserved genetic sequences between SARS-CoV and SARS-CoV-2 may be important for their stability within host cells and ability to infect and replicate efficiently. The methodology was published in Biochemical and Biophysical Research Communications.
Viral genomes are replicated in host cells through expression of viral genes, which is a key factor in viral virulence. Host cells attempt to restrict viral amplification by activating innate immune signaling pathways. Some viruses have evolved to carry RNA fragments with characteristic high-dimensional structures within their genomes to regulate the fate of viral replication in host cells through regulation of RNA degradation.
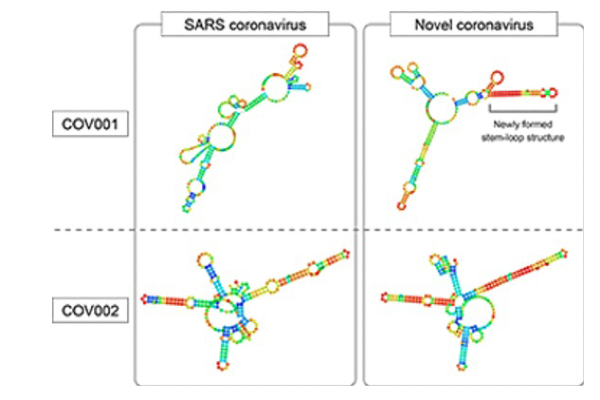
The hairpin shape of these parts of the viruses' genomes may help the viruses avoid a host's immune system. Due to a few important mutations, the genome area named COV001 does not form a hairpin structure in the SARS virus (top left) but does in the COVID-19 virus (top right). Image courtesy of Wakida et al.
Researchers developed a system called "Fate-seq" to identify RNA sequences derived from the genomes of RNA and DNA viruses that contribute to the stability of viral RNA inside cells.
"The Fate-seq technique is a very simple idea. We combined existing technologies in a new way," explained Nobuyoshi Akimitsu, PhD, a professor at the University of Tokyo Isotope Science Center.
Fate-seq consists of four steps:
- In silico design of viral sequences based on the genomic sequences of DNA and RNA viruses
- Construction of a vector library of viral sequences
- Transcription of the resulting mRNAs in vitro using the vector library as a template
- Transfection of the mRNA library into cells and measured using next-generation sequencing
These steps allow the researchers to access the genomic stability of selected viral sequences.
The researchers used Fate-seq to examine genomic conservation -- regions of identical or similar sequences within a genome -- in coronaviruses. They compared the genomes of 37 viruses belonging to the Coronaviridae family, including bat coronaviruses and SARS-CoV-2.
The analysis revealed that while there was low genomic conservation upstream and downstream, conservation near the central region was high. They identified 21 viral sequences that were abundant in the SARS-CoV genome and two sequences that exhibited a high rate of conservation (16,901-17,160 nt and 20,901-21,160 nt in the SARS-CoV genome).
When the researchers compared these two regions from SARS-CoV to SARS-CoV-2, they found that the nucleotides were highly conserved (92% and 84% similar, respectively), suggesting that these regions may play an important role in viral function in the host cell.
Next, the team predicted the secondary structure of these regions. The first region, called COV001, forms a stem-loop in SARS-CoV-2 but not in SARS-CoV. Stem and loops are short pieces of RNA that, instead of remaining in a straight line, fold forward and bind onto themselves, forming a hairpin shape. Importantly, they can provide stability to RNA structures through base pairing.
The second conserved region was predicted to form a stem-loop in both viruses. This offers an indication as to why SARS-CoV-2 may be more virulent than SARS-CoV. Both regions are almost completely conserved among the 1,116 SARS-CoV-2 genomic sequences sampled. This could enhance SARS-CoV-2 genomic stability within host cells through inhibition of the host's RNA degradation machinery and consequently improve viral replication and virulence.
"The stem-and-loop structure of this SARS-CoV-2 genetic fragment is very stable in computer models and we propose that this structure might enhance survival of the virus," Akimitsu said.
The researchers hope to use Fate-seq to understand the fundamental rules of RNA stability and advance new types of medicine.
Do you have a unique perspective on your research related to virology? Contact the editor today to learn more.
---
Copyright © 2020 scienceboard.net