


March 21, 2018 -- Here at The Science Advisory Board, we recognize the study of cells has been instrumental in understanding biological processes. Today’s advancements in cellular biology draw on single-cell analysis. This study provides insight into how single-cell analysis is shaping current research, as well as improvements fellow scientists believe could be made to the current applications and technologies.
In the past, research on genes and proteins expressed in specific types of cells has been performed on samples containing many cells, with analysis reflecting the data averaged across them. More recently, studies have indicated differences in cells of the same type or within the same population. Consequently, researchers are beginning to conduct single-cell analysis to study the unique properties of individual cells.
Individual cells have been analyzed microscopically for quite some time in order to study cell morphology and behavior, but with a growing interest in genomics and proteomics, demand for analysis of genes and proteins within single cells has increased. We have seen new developments for single-cell genomic tools, with the capability to amplify DNA and RNA or individual cells. These advances have lead to targeted DNA sequencing, miRNA analysis, and gene expression studies. Furthermore, flow cytometry techniques have led scientists to better resolve the expression of gene products in single cells. Such advances are transforming our field and improving our understanding of individual cells.
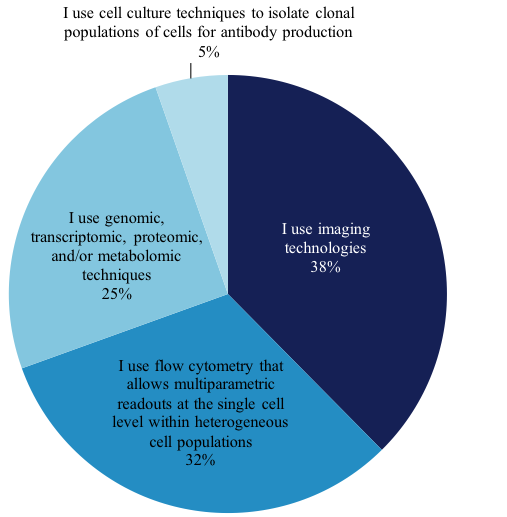
We wanted to explore this growing technique and find out from our members how they perform single-cell analysis work. Of the members that perform single-cell analysis, 1,157 answered the question, “What BEST describes the nature of the single-cell analysis work you perform?” Their answers are as follows:
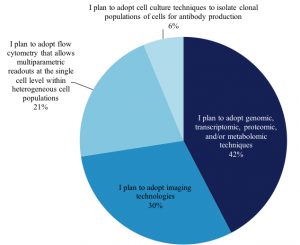
We also asked 765 scientists who plan to adopt single-cell analysis in the next 12-24 months, which techniques they plan to use, and we found the distribution of techniques remains very similar to those in use today:
Single-cell analysis encompasses the application of genomic, transcriptomic, proteomic, and/or metabolomics techniques to the analysis of isolated individually partitioned single cells. These techniques, coupled with new methods for cell isolation and partitioning, support the interrogation of single cells at increased levels of scale and throughput than previously possible.
In order to analyze a single cell, it must be reliably isolated. Scientists have found that to isolate a single cell from a heterogeneous population that preserves biological integrity can be difficult. Many continue to use tried and true methods including micromanipulation, laser-capture microdissection and fluorescent-activated cell sorting (FACS). We at the Science Advisory Board have found there is an increasing number of new products on the market to isolate single cells with greater speed and throughput.
Isolation of single cells using microfluidics coupled with transcriptome analysis or mass cytometry emphasize analysis of many single cells in a single run. We have found providers of this technology include: he microfluidics-based C1™ Single-Cell Analysis System from Fluidigm, the DEPArray™ NxT System from Menarini Silicon Biosystems, and the Chromium™ Single Cell Controller from 10x Genomics.
One of our members finds isolation to be a significant bottleneck:
“Relating the molecular analysis of an isolated cell to its tissue location [physiology]/function is critical. While downstream molecular analyses are becoming easier by the day, the very beginning, namely the isolation of cells that the researcher can confidently know where they are coming from remains the biggest bottleneck in single cell investigations.”
When studying small amounts of RNA contained in a single cell, at least a few cycles of amplification are usually necessary. Unfortunately, sources of error can creep into the workflow during the amplification process. This could happen, for example, in RNA amplification when studying single-RNA transcriptomes; the different RNA levels that result could mistakenly be interpreted as cell-to-cell differences. RNA-Seq methods that employ molecular barcodes are designed to minimize or eliminate that experimental error. Furthermore, false positive can appear during DNA amplification, because DNA polymerases are inherently error-prone. One scientist told us it is critically important to have good knowledge of the error rates associated with a particular method prior to investigating and reporting on biological variation. Many of our members hope that in the future improvements will be made to single cell molecule sequencing that will enable them to measure mutations in single cells directly and without amplification.
Scientists have found that the average RNA expression of a group of cells is not necessarily the same as the RNA expression of a single cell from the same population. Some genes are expressed continuously at a low level, while others are expressed strongly for only a brief period of time. Such patterns of expression become averaged out in macroscopic measurements – and are a significant driver of single-cell analysis – the desire to reveal and understand, at the level of a single cell, the diversity of expression that is masked in the analysis of many cells simultaneously.
An advance in RNA sequencing known as RNA-capture-seq uses tiling arrays to “zoom in” on part of the transcriptome prior to sequencing. This gives better resolution and focuses the sequencing only on the area of interest. Our members have told is Agilent Technologies, Illumina, and Roche Nimblegen as well as others offer target-enrichment tools for RNA sequencing. These target enrichment tools have been instrumental in overcoming barriers for work in single-cell analysis and are especially well suited for clinical-research applications such as circulating tumor cells or single cells biopsied from embryos. Translational researchers are also in heterogeneous tumor samples, where it can be very important to look at one cell at a time.
Our members have told us they are looking for new approaches to single-cell analyses. Scientists hope for new analytical measures and manipulations of cellular contents, structure, and activity at the single cell level significantly beyond what is currently available. We asked our members to tell us what they considered the biggest barrier to their current single-cell research. A few are listed below:
3′ bias and depth of sequencing. A lot of genomic regions of interest can be identified by single-cell sequencing, but establishing that these elements are “real” differences and that this event is a statistically significant difference for cells isolated for a given perturbation is not really easy to do and leads to a lot of false positives for regions of interest.
Another scientist noted:
Analyzing single cells using omic techniques is challenging because the loss of material causes dropouts in the sequence and because sequencing errors are difficult to distinguish from real mutations. Any “omic” technique relies on the fact that it should be unbiased and large enough to give a true global picture of what is happening at the cellular. We are really limited by the throughput to be able to sample enough cells of a tumor to demonstrate that we have been able to genomically reconstruct the entire heterogeneity of the tumor such that we can now explain and predict the evolution of the tumor based on the genomics of the cellular population.
Cost was a concern amongst many of our members:
Dissociation of heterogeneous tissues giving a decent yield of viable cells. If you want to routinely do studies that involve hundreds of thousands of cells, even at an end-to-end cost of $0.10 per cell, that translates to $10,000 to $100,000 per experiment. The cost ultimately needs to be $0.01 per cell for use of the technology to become routine by most research labs.
This is an exciting time for cell biology because according to the NIH, research based on single-cell analysis will benefit from the following solutions:
- Devices and reagents to perform novel total (i.e., “-omic”) molecular and/or functional analyses of a wide variety of cell types (e.g., imaging-based spatial “-omics”, microdroplet-based high throughput “-omics”).
- Combinations of tools for multiplex analysis and/or manipulation of single cells to maximize data content over many parameters (e.g., gene expression, protein interaction, metabolic states, electrochemical dynamics, signal secretion/reception/transduction, cell adhesive properties, intercellular communication, cytoarchitectonic or migratory changes).
- Tools that provide significant advances in assay sensitivity, selectivity, scalability, or spatiotemporal resolution of molecules/structures/activities of single cells in situ
- Automated manipulation or precise perturbation for scalable analysis of single cells, including parallel readouts in multiple cells and/or speed of processing.
- Tools and technologies that enable and transform single-cell analysis in clinical tissue biopsies.
- Systems-level single-cell dataset analysis or modeling, including computational approaches, in the context of tissues or whole organisms.
These needed technologies and tools must be developed in the context of a variety of experimental scenarios, such as:
- Identification of spatiotemporal transitions in cellular states (e.g., progenitor lineage determination, cellular aging, clonal selection and evolution, asymmetric division, cell specification, cell reprogramming).
- Detection or isolation of rare cells in a population (e.g., stem cells, tumor-initiating or metastatic cells, drug resistant cells).
- Elucidation of the cell molecular signatures at levels including DNA, RNA, protein, or metabolite) or functional consequences of molecular changes (e.g., genomic stability, epigenetic regulation, RNA modification or editing, protein modification or interaction, lipid metabolism)
- Characterization of heterogeneous cell responses to environmental changes (e.g., homeostatic perturbation, modulation of niche/microenvironment, morphogens or cell-to-cell signaling, toxic exposure, experience-dependent plasticity, host cell responses to infectious, immunological or allergic challenges).
Does your research involve single-cell analysis? What is the nature of your single cell analysis work? Join the discussion below!
Copyright © 2018 scienceboard.net