



March 4, 2021 -- Researchers have identified protein compounds that are potent inhibitors of the SARS-CoV-2 main protein, a key enzyme that enables the replication of the deadly virus. Their screening and synthetic chemistry approach was detailed in an article published March 3 in Angewandte Chemie.
SARS-CoV-2 encodes two overlapping polyproteins that produce the main protease. Subsequent processing of the main protease is critical for the assembly of the viral replication-transcription complex, the release of functional viral proteins, and ultimately replication of SARS-CoV-2.
The main protease forms a homodimer through a covalent mechanism that can be utilized for the design of targeted inhibitors bearing an electrophilic warhead.
The enzyme has a unique substrate specificity mainly recognizing residues ranging from the cleavage site at position 4 (P4) to P1' and processing a preference for glutamine at the P1 position. Because this specificity does not exist in human proteases, pharmacists at the University of Bonn hypothesized that it is feasible to develop selective protease inhibitors without toxicities in patients.
"The main protease is an extremely promising starting point for coronavirus drug research," said Christa Müller, PhD, group leader at the Pharmaceutical Institute at the University of Bonn, in a statement. "If this enzyme is blocked, viral replication in the body's cells is stopped."
Main protease fluorescent bioassay
The researchers developed a new SARS-CoV-2 main protease assay for quantitative high-throughput screening. They began by establishing a bacterial expression system for the SARS-CoV-2 main protease where BL21 Escherichia coli cells were transformed with a DNA construct encoding the protease with an N-terminally fused main protease cleavage site and a C-terminal histidine tag.
The tag was used to monitor the proteolytic activity of the protease in combination with an internally quenched fluorescent peptide substrate. When the main protease is cleaved, the fluorophore produces a measurable fluorescent signal. However, if an inhibitor of the main protease is simultaneously administered, the fluorophore and resulting fluorescence is blocked.
"A suitable inhibitor must bind sufficiently tightly to the main protease to be able to block its active site," explained Michael Gütschow, PhD, who heads an independent research group on such inhibitors at the Pharmaceutical Institute of the University of Bonn.
Finding and building potent main protease inhibitors
Focused libraries were compiled taking inhibitors of related cysteine proteases and cysteine-reactive compounds into account. These initial libraries included chloroacetamide derivatives with potential electrophilic reactivity toward the active site of the cysteine nucleophile of the main protease and Michael acceptors (weak irreversible inhibitors of the protease).
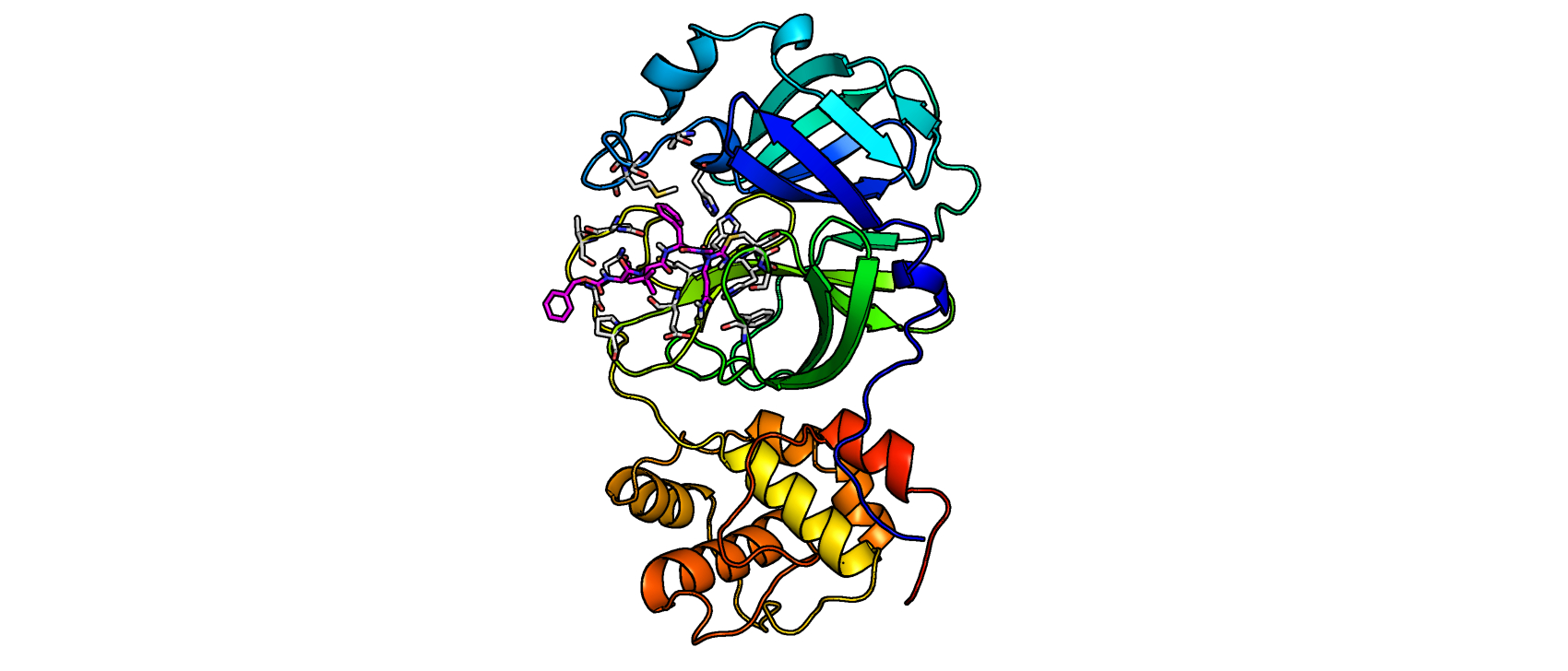
The SARS-CoV-2 main protease with one of the newly developed inhibitors in the active center. The individual domains of the protein are shown in different colors, the inhibitor in pink. Image courtesy of V. Namasivayam/Pharmazeutisches Institut/Uni Bonn.
In a separate library of peptide nitriles, the researchers discovered several hits of mostly azapeptide nitriles in which the cyano group was connected to a nitrogen replacing the P1 the carbon group.
"For most of the test compounds, we observed no enzyme inhibition," reported Gütschow. "But on rare occasions in our comprehensive tests, fluorescence was suppressed. These were the hits we had hoped for in our search for inhibitors of the viral protease."
Based on the results from the high-throughput screening method, the researchers chose to design azadipeptide nitriles with enhanced features to increase their effectiveness at inhibiting the main protease. They synthetically designed peptidomimetics (small protein-like chains designed to mimic a peptide) candidates -- azadipeptide nitriles with glutamine side chains and some with additional amino acids.
They found that the structural expansion (those with additional amino acids) improved the biological activity of those compounds through specific noncovalent interactions of the P1-P4 residues with the active-site subsites.
To further characterize the inhibitors, the researchers performed a covalent docking using the x-ray crystal structure of the SARS-CoV-2 main protease in complex with a peptidic acrylate inhibitor. Through virtual screening of compound libraries, the team identified indole esters that were extremely potent inhibitors of the main protease. These small molecular fragments have a high ligand efficiency and can serve as excellent scaffolds for drug development.
The researchers also investigated the potent compounds for their ability to not only block the viral main protease, but also cathepsin L -- a human lysosomal cysteine protease that contributes to the activation of the SARS-CoV-2 spike protein. They found that the indole esters did not inhibit cathepsin L while the azapeptide nitriles exhibited strong anti-cathepsin L activity.
"Some of the compounds even have another effect," Müller said. "They also inhibit a human enzyme that helps the virus enter body cells."
While clinical trials have yet to prove whether the candidates effectively inhibit replication of SARS-CoV-2 in humans, "The best compounds represent promising lead structures for drug development," according to Müller.
Do you have a unique perspective on your research related to infectious diseases or chemistry? Contact the editor today to learn more.
---




Copyright © 2021 scienceboard.net
Member Rewards
Earn points for contributing to market research. Redeem your points for merchandise, travel, or even to help your favorite charity.

Research Topics
Interact with an engaged, global community of your peers who come together to discuss their work and opportunities.
Conferences
Connect


